

This is a preprint.
Identification of non-canonical peptides with moPepGen
- PMID: 38585946
- PMCID: PMC10996593
- DOI: 10.1101/2024.03.28.587261
Identification of non-canonical peptides with moPepGen
Update in
-
Identification of non-canonical peptides with moPepGen.Nat Biotechnol. 2025 Jun 16. doi: 10.1038/s41587-025-02701-0. Online ahead of print. Nat Biotechnol. 2025. PMID: 40523945
Abstract
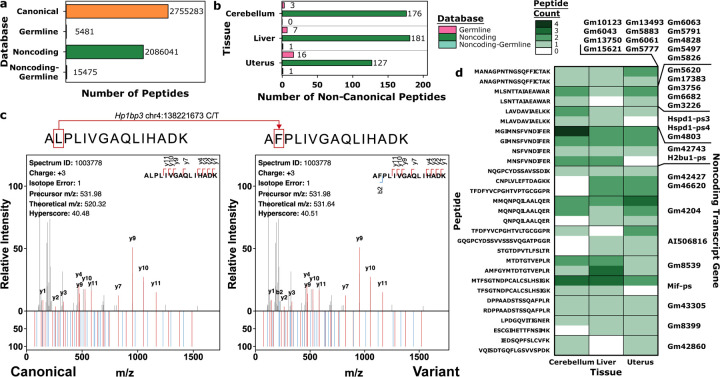
Proteogenomics is limited by challenges of modeling the complexities of gene expression. We create moPepGen, a graph-based algorithm that comprehensively generates non-canonical peptides in linear time. moPepGen works with multiple technologies, in multiple species and on all types of genetic and transcriptomic data. In human cancer proteomes, it enumerates previously unobservable noncanonical peptides arising from germline and somatic genomic variants, noncoding open reading frames, RNA fusions and RNA circularization.
Conflict of interest statement
Conflicts of Interest PCB sits on the Scientific Advisory Boards of Intersect Diagnostics Inc., and previously sat on those of Sage Bionetworks and BioSymetrics Inc. All other authors declare no conflicts of interest.
Figures










References
Online Methods References
-
- Zhu C., Liu L. Y., Kislinger T. & Boutros P. C. call-NonCanonicalPeptide: nextflow pipeline to generate custom databases of non-canonical peptides for proteogenomic analysis, Source code. https://github.com/uclahs-cds/pipeline-call-NonCanonicalPeptide (2025).
-
- Di Tommaso P. et al. Nextflow enables reproducible computational workflows. Nat Biotechnol 35, 316–319 (2017). - PubMed
-
- Patel Y. et al. Metapipeline-DNA: A Comprehensive Germline & Somatic Genomics Nextflow Pipeline. bioRxiv 2024.09.04.611267 (2024) doi: 10.1101/2024.09.04.611267. - DOI
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources