

This is a preprint.
Mutational scanning of CRX classifies clinical variants and reveals biochemical properties of the transcriptional effector domain
- PMID: 38585983
- PMCID: PMC10996540
- DOI: 10.1101/2024.03.21.585809
Mutational scanning of CRX classifies clinical variants and reveals biochemical properties of the transcriptional effector domain
Update in
-
Mutational scanning of CRX classifies clinical variants and reveals biochemical properties of the transcriptional effector domain.Genome Res. 2024 Oct 29;34(10):1540-1552. doi: 10.1101/gr.279415.124. Genome Res. 2024. PMID: 39322280 Free PMC article.
Abstract
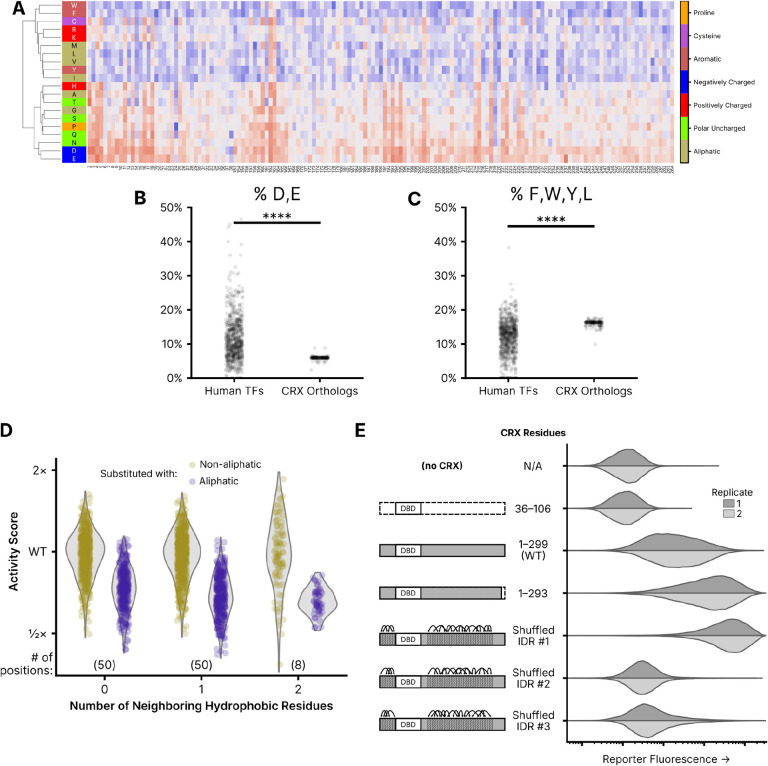
Cone-Rod Homeobox, encoded by CRX, is a transcription factor (TF) essential for the terminal differentiation and maintenance of mammalian photoreceptors. Structurally, CRX comprises an ordered DNA-binding homeodomain and an intrinsically disordered transcriptional effector domain. Although a handful of human variants in CRX have been shown to cause several different degenerative retinopathies with varying cone and rod predominance, as with most human disease genes the vast majority of observed CRX genetic variants are uncharacterized variants of uncertain significance (VUS). We performed a deep mutational scan (DMS) of nearly all possible single amino acid substitution variants in CRX, using an engineered cell-based transcriptional reporter assay. We measured the ability of each CRX missense variant to transactivate a synthetic fluorescent reporter construct in a pooled fluorescence-activated cell sorting assay and compared the activation strength of each variant to that of wild-type CRX to compute an activity score, identifying thousands of variants with altered transcriptional activity. We calculated a statistical confidence for each activity score derived from multiple independent measurements of each variant marked by unique sequence barcodes, curating a high-confidence list of nearly 2,000 variants with significantly altered transcriptional activity compared to wild-type CRX. We evaluated the performance of the DMS assay as a clinical variant classification tool using gold-standard classified human variants from ClinVar, and determined that activity scores could be used to identify pathogenic variants with high specificity. That this performance could be achieved using a synthetic reporter assay in a foreign cell type, even for a highly cell type-specific TF like CRX, suggests that this approach shows promise for DMS of other TFs that function in cell types that are not easily accessible. Per-position average activity scores closely aligned to a predicted structure of the ordered homeodomain and demonstrated position-specific residue requirements. The intrinsically disordered transcriptional effector domain, by contrast, displayed a qualitatively different pattern of substitution effects, following compositional constraints without specific residue position requirements in the peptide chain. The observed compositional constraints of the effector domain were consistent with the acidic exposure model of transcriptional activation. Together, the results of the CRX DMS identify molecular features of the CRX effector domain and demonstrate clinical utility for variant classification.
Keywords: CRX; acidic exposure model; deep mutational scan; intrinsically disordered domain; retina; retinopathy; transcription factor.
Conflict of interest statement
Declaration of Interests B.A.C. is on the scientific advisory board of Patch Biosciences. The other authors declare no competing interests.
Figures
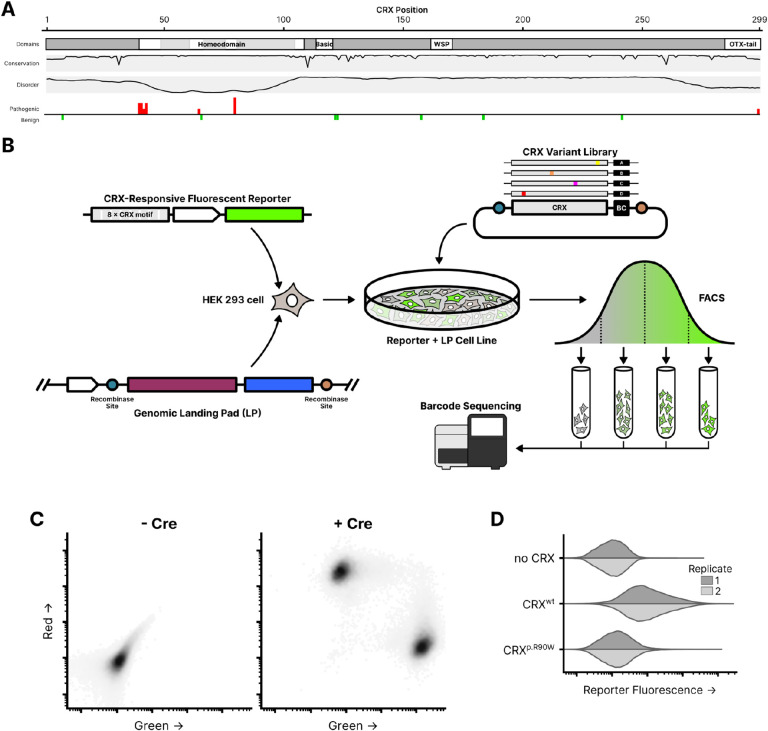
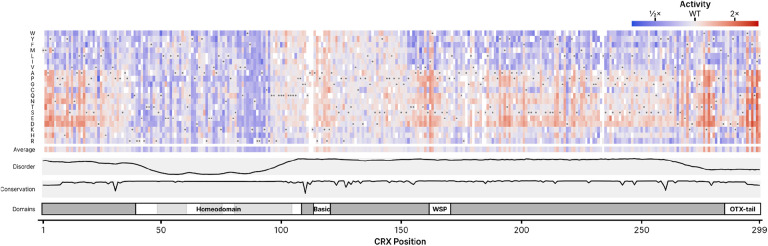
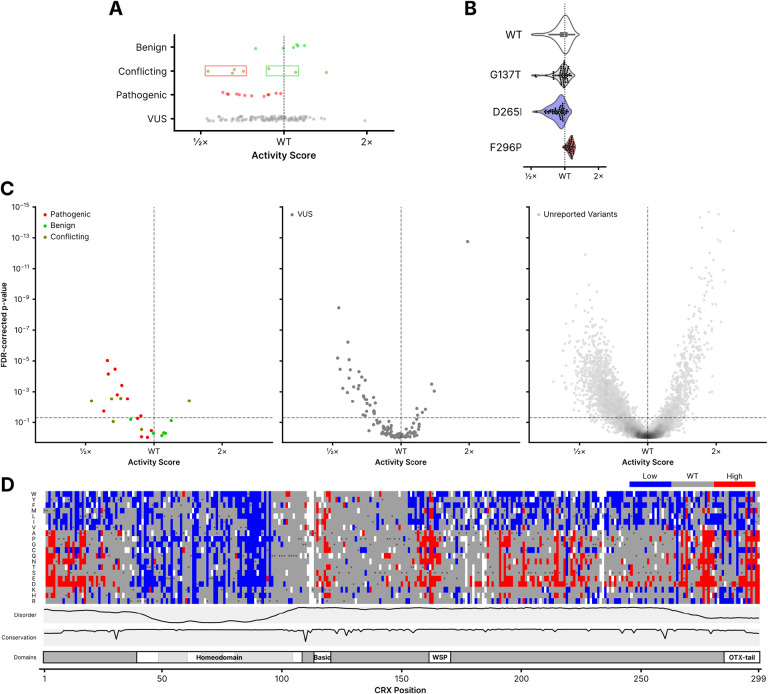
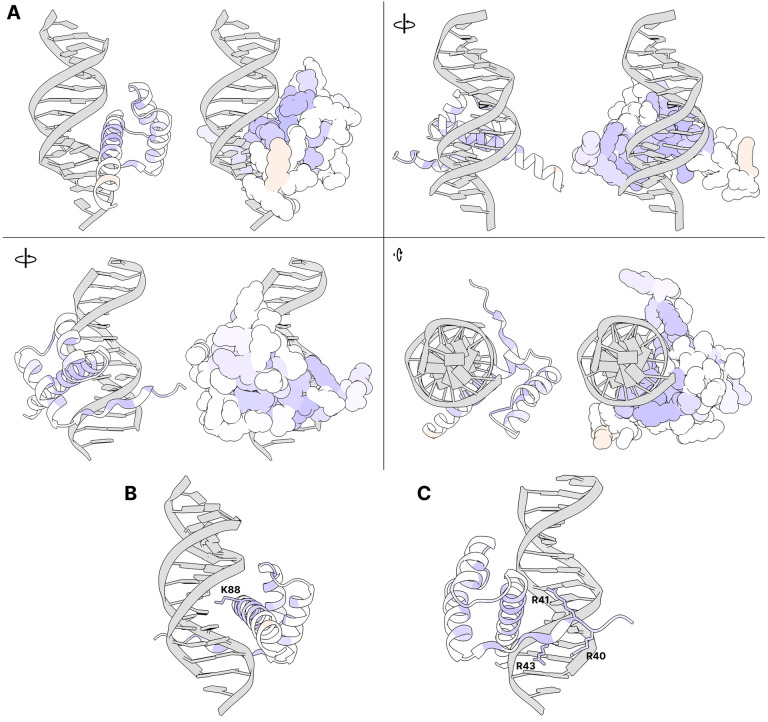
References
-
- Brnich SE, Abou Tayoun AN, Couch FJ, Cutting GR, Greenblatt MS, Heinen CD, Kanavy DM, Luo X, McNulty SM, Starita LM, et al. 2020. Recommendations for application of the functional evidence PS3/BS3 criterion using the ACMG/AMP sequence variant interpretation framework. Genome Medicine 12: 3. - PMC - PubMed
-
- Chen S, Wang Q-L, Xu S, Liu I, Li LY, Wang Y, Zack DJ. 2002. Functional analysis of cone–rod homeobox (CRX) mutations associated with retinal dystrophy. Human Molecular Genetics 11: 873–884. - PubMed
-
- Cheng J, Novati G, Pan J, Bycroft C, Žemgulytė A, Applebaum T, Pritzel A, Wong LH, Zielinski M, Sargeant T, et al. 2023. Accurate proteome-wide missense variant effect prediction with AlphaMissense. Science (New York, NY) 381: eadg7492. - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous