

This is a preprint.
Accelerated Bayesian inference of population size history from recombining sequence data
- PMID: 38585997
- PMCID: PMC10996539
- DOI: 10.1101/2024.03.25.586640
Accelerated Bayesian inference of population size history from recombining sequence data
Abstract
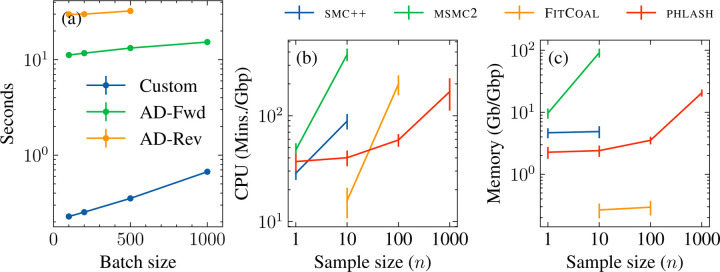
I present PHLASH, a new Bayesian method for inferring population history from whole genome sequence data. PHLASH is population history learning by averaging sampled histories: it works by drawing random, low-dimensional projections of the coalescent intensity function from the posterior distribution of a PSMC-like model, and averaging them together to form an accurate and adaptive size history estimator. On simulated data, PHLASH tends to be faster and have lower error than several competing methods including SMC++, MSMC2, and FITCOAL. Moreover, it provides a full posterior distribution over population size history, leading to automatic uncertainty quantification of the point estimates, as well to new Bayesian testing procedures for detecting population structure and ancient bottlenecks. On the technical side, the key advance is a novel algorithm for computing the score function (gradient of the log-likelihood) of a coalescent hidden Markov model: when there are hidden states, the algorithm requires time and memory per decoded position, the same cost as evaluating the log-likelihood itself using the naïve forward algorithm. This algorithm is combined with a hand-tuned implementation that fully leverages the power of modern GPU hardware, and the entire method has been released as an easy-to-use Python software package.
Figures














Similar articles
-
Exact Decoding of a Sequentially Markov Coalescent Model in Genetics.J Am Stat Assoc. 2024;119(547):2242-2255. doi: 10.1080/01621459.2023.2252570. Epub 2023 Oct 3. J Am Stat Assoc. 2024. PMID: 39323740 Free PMC article.
-
Decoding coalescent hidden Markov models in linear time.Res Comput Mol Biol. 2014;8394:100-114. doi: 10.1007/978-3-319-05269-4_8. Res Comput Mol Biol. 2014. PMID: 25340178 Free PMC article.
-
Robust inference of population size histories from genomic sequencing data.PLoS Comput Biol. 2022 Sep 16;18(9):e1010419. doi: 10.1371/journal.pcbi.1010419. eCollection 2022 Sep. PLoS Comput Biol. 2022. PMID: 36112715 Free PMC article.
-
Beta-PSMC: uncovering more detailed population history using beta distribution.BMC Genomics. 2022 Nov 30;23(1):785. doi: 10.1186/s12864-022-09021-6. BMC Genomics. 2022. PMID: 36451098 Free PMC article.
-
Estimating variable effective population sizes from multiple genomes: a sequentially markov conditional sampling distribution approach.Genetics. 2013 Jul;194(3):647-62. doi: 10.1534/genetics.112.149096. Epub 2013 Apr 22. Genetics. 2013. PMID: 23608192 Free PMC article.
References
-
- Baharian Soheil and Gravel Simon (2018). “On the decidability of population size histories from finite allele frequency spectra”. In: Theoretical Population Biology 120, pp. 42–51. - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources