

Treatment with ataluren in four symptomatic Duchenne carriers. A pilot study
- PMID: 38586166
- PMCID: PMC10997039
- DOI: 10.36185/2532-1900-398
Treatment with ataluren in four symptomatic Duchenne carriers. A pilot study
Abstract
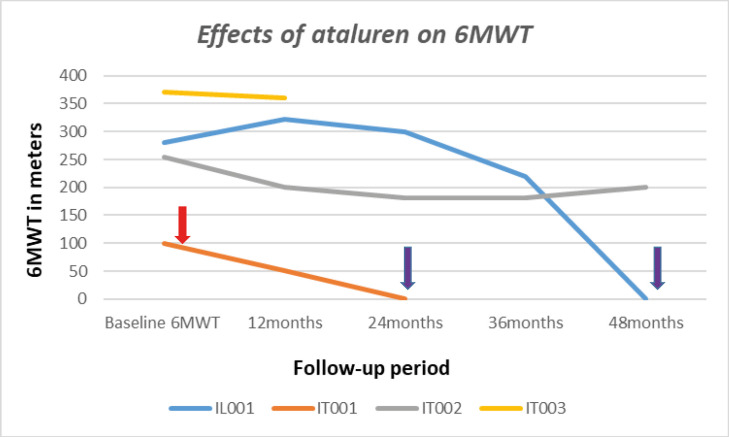
Duchenne muscular dystrophy (DMD) is a devastating X-linked neuromuscular disorder caused by dystrophin gene deletions (75%), duplications (15-20%) and point mutations (5-10%), a small portion of which are nonsense mutations. Women carrying dystrophin gene mutations are commonly unaffected because the wild X allele may produce a sufficient amount of the dystrophin protein. However, approximately 8-10% of them may experience muscle symptoms and 50% of those over 40 years develop cardiomyopathy. The presence of symptoms defines the individual as an affected "symptomatic or manifesting carrier". Though there is no effective cure for DMD, therapies are available to slow the decline of muscle strength and delay the onset and progression of cardiac and respiratory impairment. These include ataluren for patients with nonsense mutations, and antisense oligonucleotides therapies, for patients with specific deletions. Symptomatic DMD female carriers are not included in these indications and little data documenting their management, often entrusted to the discretion of individual doctors, is present in the literature. In this article, we report the clinical and instrumental outcomes of four symptomatic DMD carriers, aged between 26 and 45 years, who were treated with ataluren for 21 to 73 months (average 47.3), and annually evaluated for muscle strength, respiratory and cardiological function. Two patients retain independent ambulation at ages 33 and 45, respectively. None of them developed respiratory involvement or cardiomyopathy. No clinical adverse effects or relevant abnormalities in routine laboratory values, were observed.
Keywords: DMD symptomatic/affected carriers; Duchenne muscular dystrophy; ataluren; manifesting carriers; nonsense mutations.
©2024 Gaetano Conte Academy - Mediterranean Society of Myology, Naples, Italy.
Conflict of interest statement
The Authors have no conflicts of interest to declare that are relevant to the content of this article.
Figures
References
-
- Carter JC, Sheehan DW, Prochoroff A, et al. Muscular Dystrophies. Clin Chest Med. 2018;39(2):377-89. https://doi.org/10.1016/j.ccm.2018.01.004. 10.1016/j.ccm.2018.01.004 - DOI - PubMed
-
- Monaco AP. Dystrophin, the protein product of the Duchenne/Becker muscular dystrophy gene. Trends Biochem Sci. 1989;14(10):412-5. https://doi.org/10.1016/0968-0004(89)90290-9. 10.1016/0968-0004(89)90290-9 - DOI - PubMed
-
- Kamdar F, Garry DJ. Dystrophin-Deficient Cardiomyopathy. J Am Coll Cardiol. 2016;67(21):2533-46. https://doi.org/10.1016/j.jacc.2016.02.081. 10.1016/j.jacc.2016.02.081 - DOI - PubMed
-
- Aartsma-Rus A, Ginjaar IB, Bushby K. The importance of genetic diagnosis for Duchenne muscular dystrophy. J Med Genet. 2016;53:145-151. https://doi.org/10.1136/jmedgenet-2015-103387 10.1136/jmedgenet-2015-103387 - DOI - PMC - PubMed
-
- Mercier S, Toutain A, Toussaint A, Genetic and clinical specificity of 26 symptomatic carriers for dystrophinopathies at pediatric age. Eur J Hum Genet. 2013;21(8):855-63. https://doi.org/10.1038/ejhg.2012.269. 10.1038/ejhg.2012.269 - DOI - PMC - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical