

Impaired malin expression and interaction with partner proteins in Lafora disease
- PMID: 38588813
- PMCID: PMC11063907
- DOI: 10.1016/j.jbc.2024.107271
Impaired malin expression and interaction with partner proteins in Lafora disease
Abstract
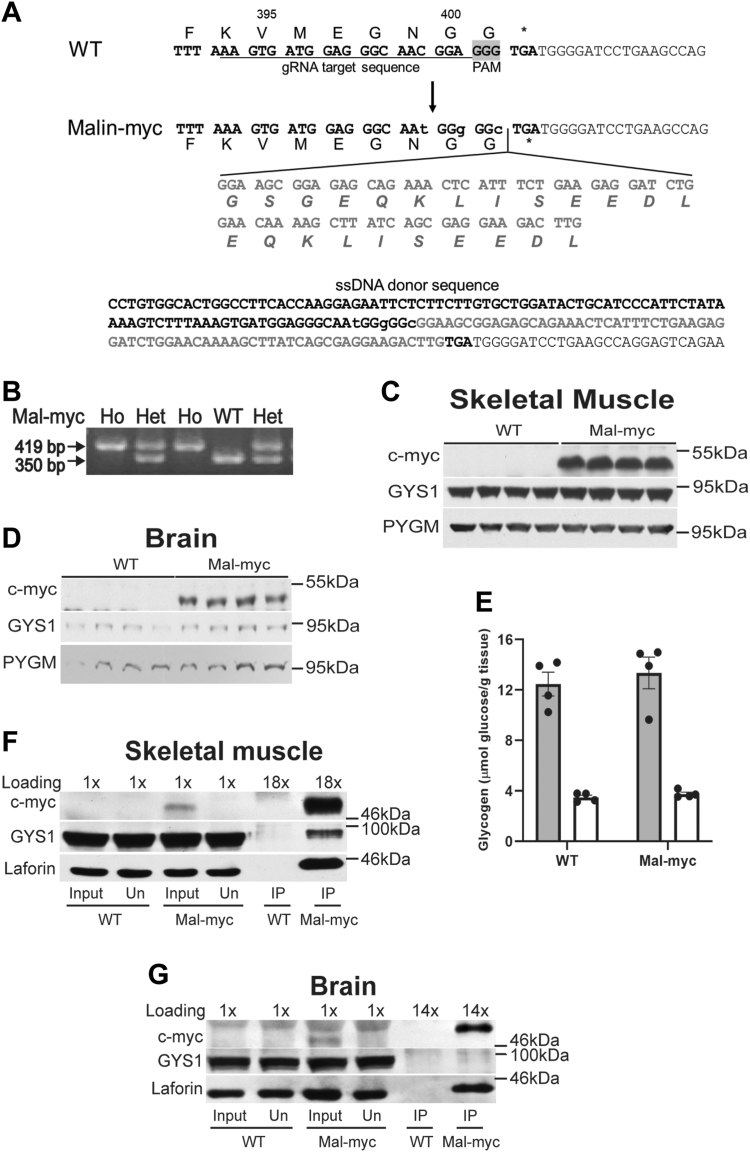
Lafora disease (LD) is an autosomal recessive myoclonus epilepsy with onset in the teenage years leading to death within a decade of onset. LD is characterized by the overaccumulation of hyperphosphorylated, poorly branched, insoluble, glycogen-like polymers called Lafora bodies. The disease is caused by mutations in either EPM2A, encoding laforin, a dual specificity phosphatase that dephosphorylates glycogen, or EMP2B, encoding malin, an E3-ubiquitin ligase. While glycogen is a widely accepted laforin substrate, substrates for malin have been difficult to identify partly due to the lack of malin antibodies able to detect malin in vivo. Here we describe a mouse model in which the malin gene is modified at the C-terminus to contain the c-myc tag sequence, making an expression of malin-myc readily detectable. Mass spectrometry analyses of immunoprecipitates using c-myc tag antibodies demonstrate that malin interacts with laforin and several glycogen-metabolizing enzymes. To investigate the role of laforin in these interactions we analyzed two additional mouse models: malin-myc/laforin knockout and malin-myc/LaforinCS, where laforin was either absent or the catalytic Cys was genomically mutated to Ser, respectively. The interaction of malin with partner proteins requires laforin but is not dependent on its catalytic activity or the presence of glycogen. Overall, the results demonstrate that laforin and malin form a complex in vivo, which stabilizes malin and enhances interaction with partner proteins to facilitate normal glycogen metabolism. They also provide insights into the development of LD and the rescue of the disease by the catalytically inactive phosphatase.
Keywords: Lafora disease; glycogen; glycogen metabolism; glycogen storage disease; laforin; malin; phosphatases.
Copyright © 2024 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Conflict of interest The authors declare the following financial interests/personal relationships which may be considered as potential competing interests: T. D. H., A. A. D.-R., and P. J. R. were consultants for Maze Therapeutics. T. D. H. is also a consultant for SAJE Pharma. The other authors declare that they have no competing interest. None of the work in this article was supported by these entities.
Figures








References
-
- Minassian B.A. Lafora's disease: towards a clinical, pathologic, and molecular synthesis. Pediatr. Neurol. 2001;25:21–29. - PubMed
-
- Ganesh S., Puri R., Singh S., Mittal S., Dubey D. Recent advances in the molecular basis of Lafora's progressive myoclonus epilepsy. J. Hum. Genet. 2006;51:1–8. - PubMed
-
- Delgado-Escueta A.V. Advances in lafora progressive myoclonus epilepsy. Curr. Neurol. Neurosci. Rep. 2007;7:428–433. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
