

Transcription bodies regulate gene expression by sequestering CDK9
- PMID: 38589534
- PMCID: PMC11021188
- DOI: 10.1038/s41556-024-01389-9
Transcription bodies regulate gene expression by sequestering CDK9
Abstract
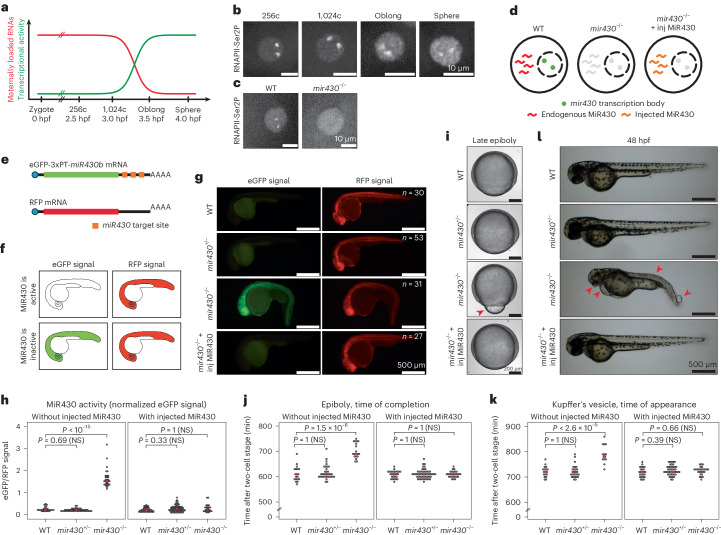
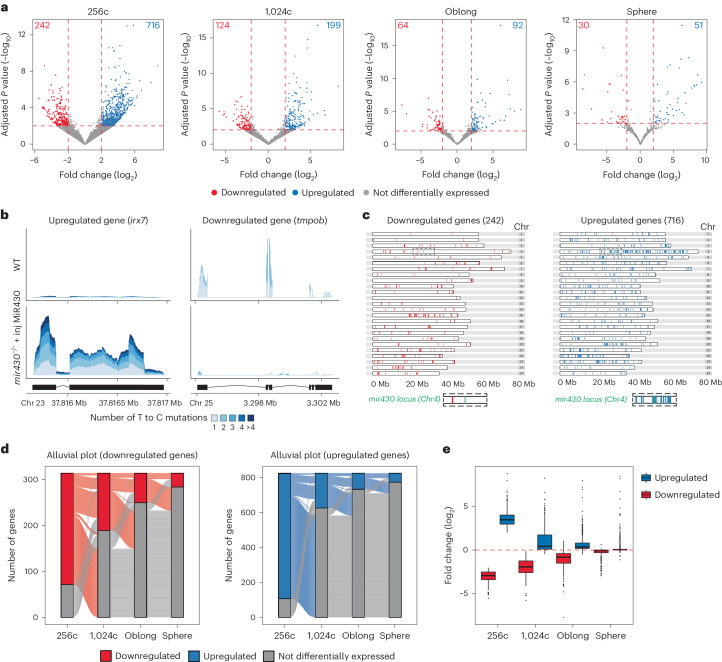
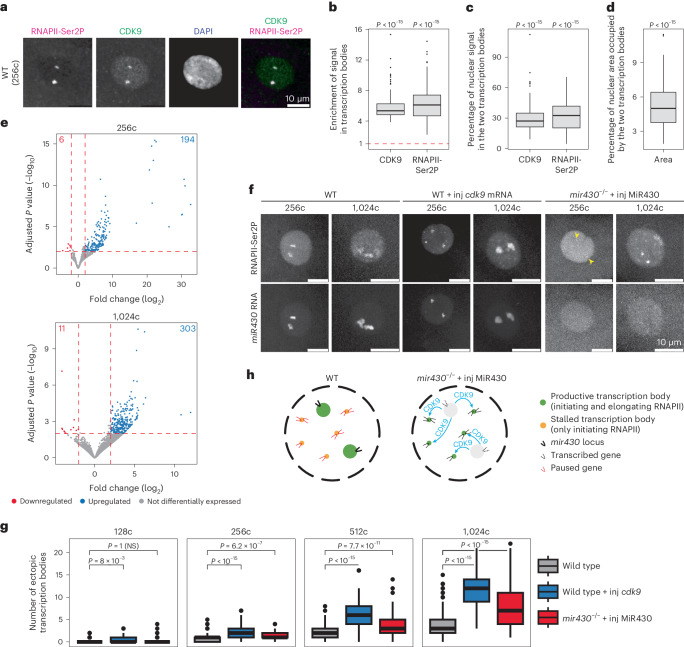
The localization of transcriptional activity in specialized transcription bodies is a hallmark of gene expression in eukaryotic cells. It remains unclear, however, if and how transcription bodies affect gene expression. Here we disrupted the formation of two prominent endogenous transcription bodies that mark the onset of zygotic transcription in zebrafish embryos and analysed the effect on gene expression using enriched SLAM-seq and live-cell imaging. We find that the disruption of transcription bodies results in the misregulation of hundreds of genes. Here we focus on genes that are upregulated. These genes have accessible chromatin and are poised to be transcribed in the presence of the two transcription bodies, but they do not go into elongation. Live-cell imaging shows that disruption of the two large transcription bodies enables these poised genes to be transcribed in ectopic transcription bodies, suggesting that the large transcription bodies sequester a pause release factor. Supporting this hypothesis, we find that CDK9-the kinase that releases paused polymerase II-is highly enriched in the two large transcription bodies. Overexpression of CDK9 in wild-type embryos results in the formation of ectopic transcription bodies and thus phenocopies the removal of the two large transcription bodies. Taken together, our results show that transcription bodies regulate transcription by sequestering machinery, thereby preventing genes elsewhere in the nucleus from being transcribed.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures













References
Publication types
MeSH terms
Substances
Grants and funding
- 101003023/EC | EU Framework Programme for Research and Innovation H2020 | H2020 Priority Excellent Science | H2020 European Research Council (H2020 Excellent Science - European Research Council)
- 94773/Volkswagen Foundation (VolkswagenStiftung)
- VA 1209/2-1/Deutsche Forschungsgemeinschaft (German Research Foundation)
- na/Max-Planck-Gesellschaft (Max Planck Society)
- na/Université de Lausanne (University of Lausanne)
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
