

Concurrent inhibition of oncogenic and wild-type RAS-GTP for cancer therapy
- PMID: 38589574
- PMCID: PMC11111408
- DOI: 10.1038/s41586-024-07205-6
Concurrent inhibition of oncogenic and wild-type RAS-GTP for cancer therapy
Abstract
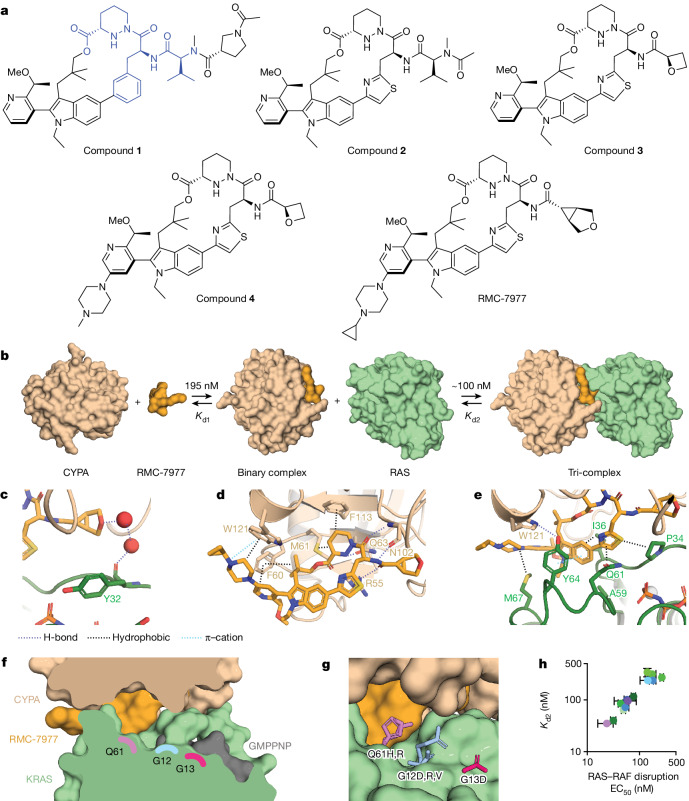
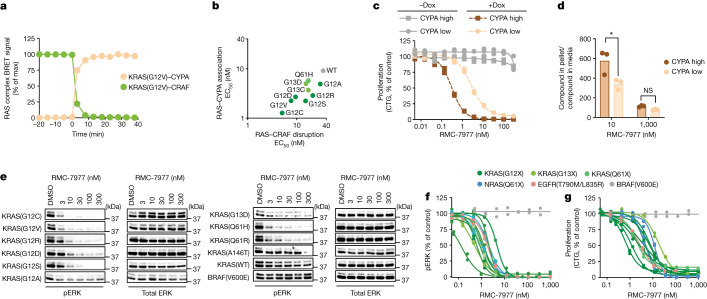
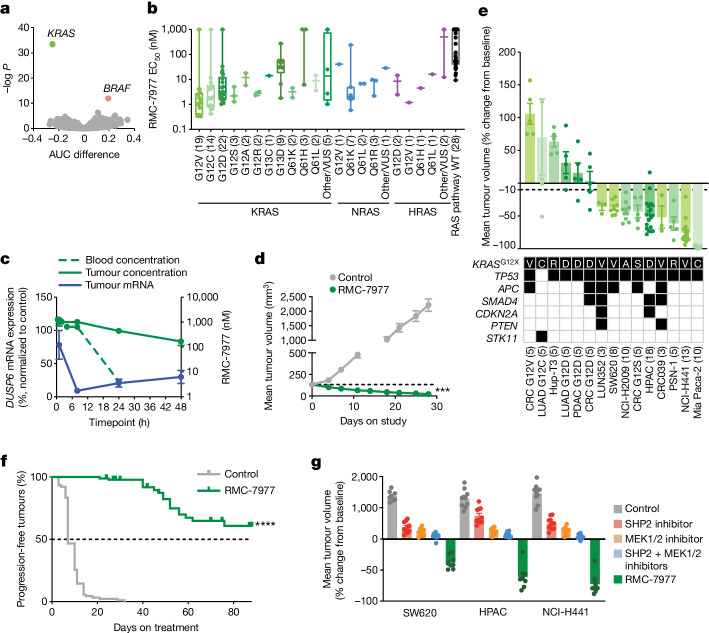
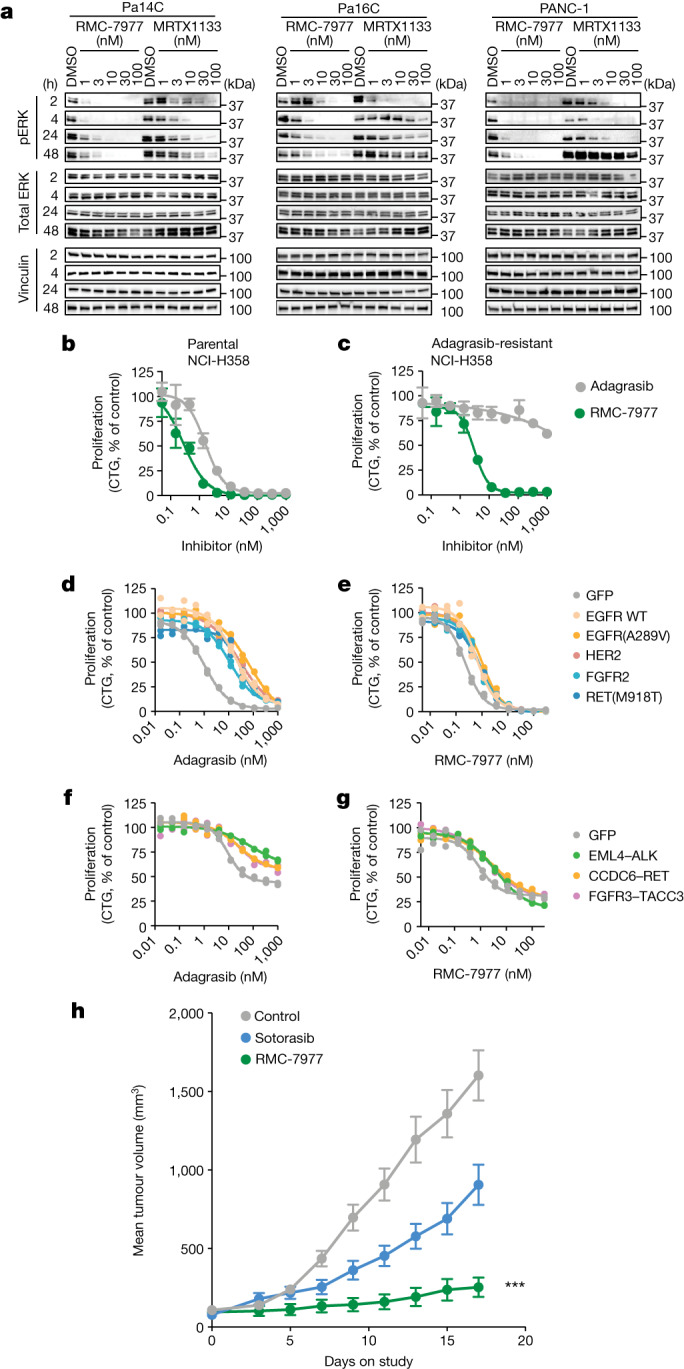
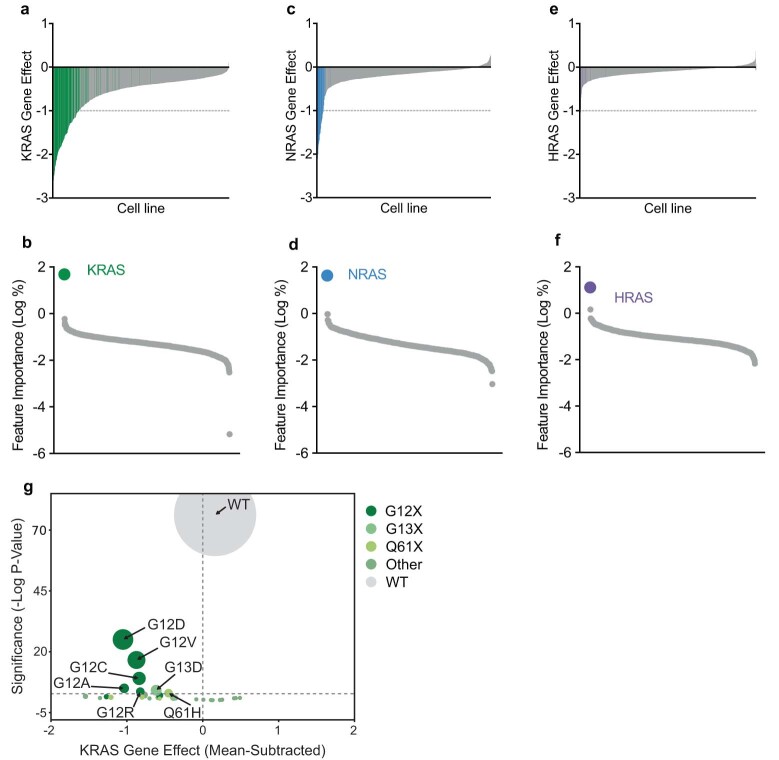
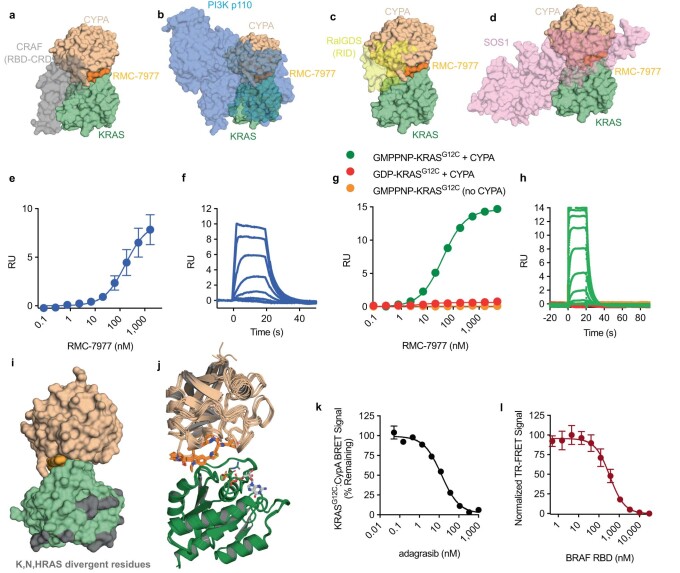
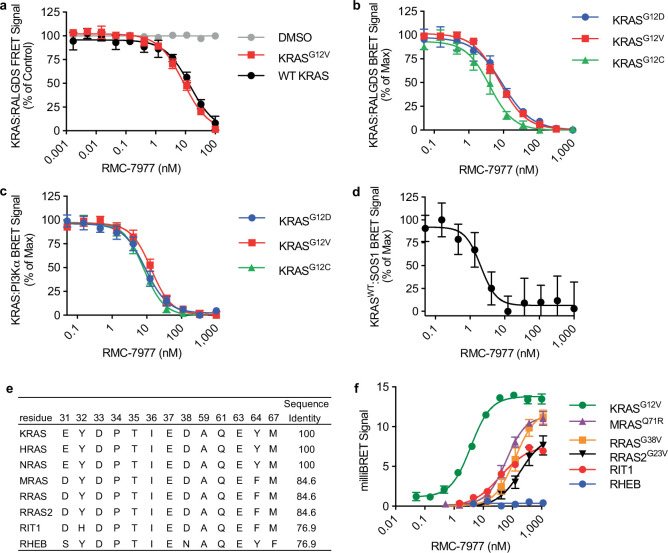
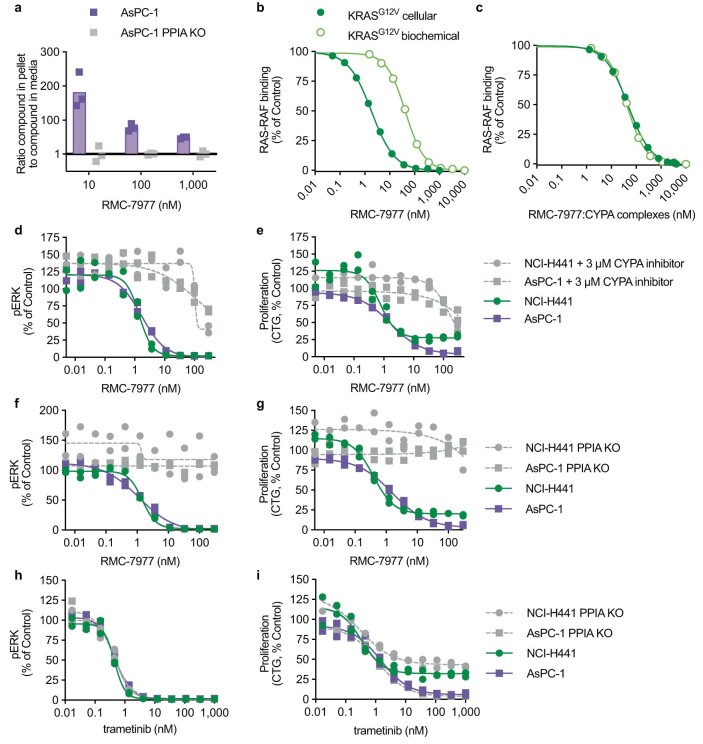
RAS oncogenes (collectively NRAS, HRAS and especially KRAS) are among the most frequently mutated genes in cancer, with common driver mutations occurring at codons 12, 13 and 611. Small molecule inhibitors of the KRAS(G12C) oncoprotein have demonstrated clinical efficacy in patients with multiple cancer types and have led to regulatory approvals for the treatment of non-small cell lung cancer2,3. Nevertheless, KRASG12C mutations account for only around 15% of KRAS-mutated cancers4,5, and there are no approved KRAS inhibitors for the majority of patients with tumours containing other common KRAS mutations. Here we describe RMC-7977, a reversible, tri-complex RAS inhibitor with broad-spectrum activity for the active state of both mutant and wild-type KRAS, NRAS and HRAS variants (a RAS(ON) multi-selective inhibitor). Preclinically, RMC-7977 demonstrated potent activity against RAS-addicted tumours carrying various RAS genotypes, particularly against cancer models with KRAS codon 12 mutations (KRASG12X). Treatment with RMC-7977 led to tumour regression and was well tolerated in diverse RAS-addicted preclinical cancer models. Additionally, RMC-7977 inhibited the growth of KRASG12C cancer models that are resistant to KRAS(G12C) inhibitors owing to restoration of RAS pathway signalling. Thus, RAS(ON) multi-selective inhibitors can target multiple oncogenic and wild-type RAS isoforms and have the potential to treat a wide range of RAS-addicted cancers with high unmet clinical need. A related RAS(ON) multi-selective inhibitor, RMC-6236, is currently under clinical evaluation in patients with KRAS-mutant solid tumours (ClinicalTrials.gov identifier: NCT05379985).
© 2024. The Author(s).
Conflict of interest statement
P.L. reports grants to his institution from Amgen, Mirati, Revolution Medicines, Boehringer Ingelheim and Virtec Pharmaceuticals. P.L. reports consulting fees or honoraria from Black Diamond Therapeutics, AmMax, OrbiMed, PAQ-Tx, Repare Therapeutics, Boehringer Ingelheim and Revolution Medicines, as well as membership on the Scientific Advisory Board of Frontier Medicines, Ikena, Biotheryx and PAQ-Tx (consulting fees and equity in each). A.J.A. has consulted for Anji Pharmaceuticals, Affini-T Therapeutics, Arrakis Therapeutics, AstraZeneca, Boehringer Ingelheim, Oncorus, Merck & Co., Mirati Therapeutics, Nimbus Therapeutics, Plexium, Revolution Medicines, Reactive Biosciences, Riva Therapeutics, Servier Pharmaceuticals, Syros Pharmaceuticals, T-knife Therapeutics, Third Rock Ventures, and Ventus Therapeutics. A.J.A. holds equity in Riva Therapeutics. C.J.D. has consulted or been an advisory board member for SKY Therapeutics, Deciphera Pharmaceuticals, Kestral Therapeutics, Mirati Therapeutics, Reactive Biosciences, Revere Pharmaceuticals, Revolution Medicines, SHY Therapeutics and Sanofi. C.A. has received research fees from Revolution Medicines, Aelin Therapeutics, Verastem, Roche and Boehringer Ingelheim. The other authors declare no competing interests.
Figures
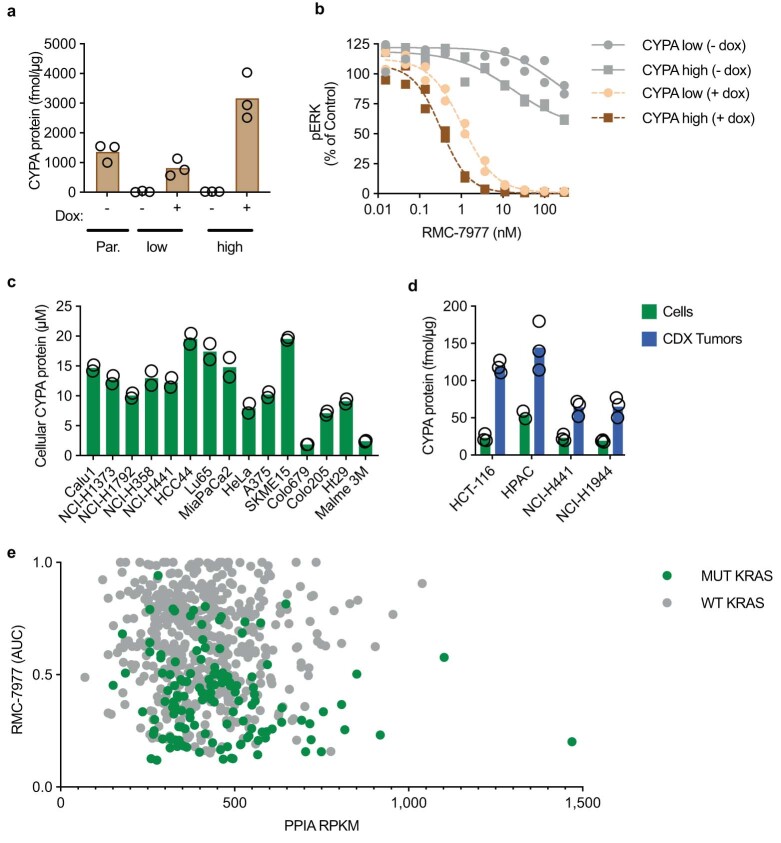
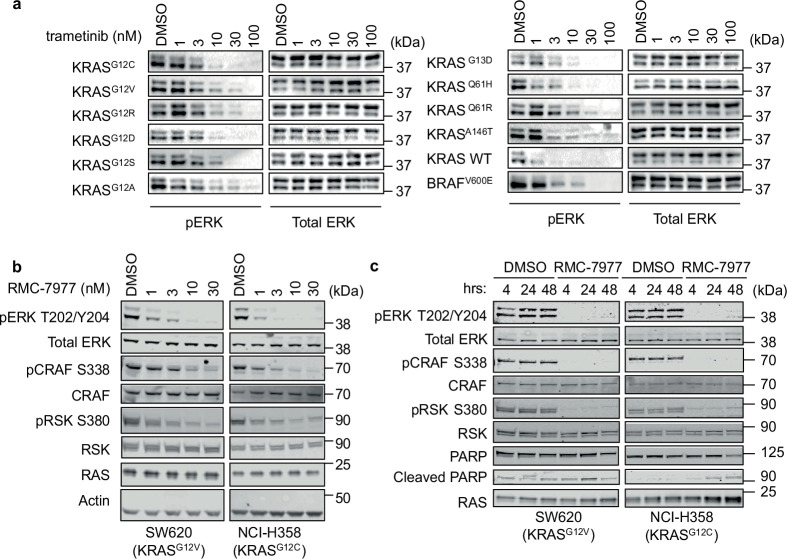
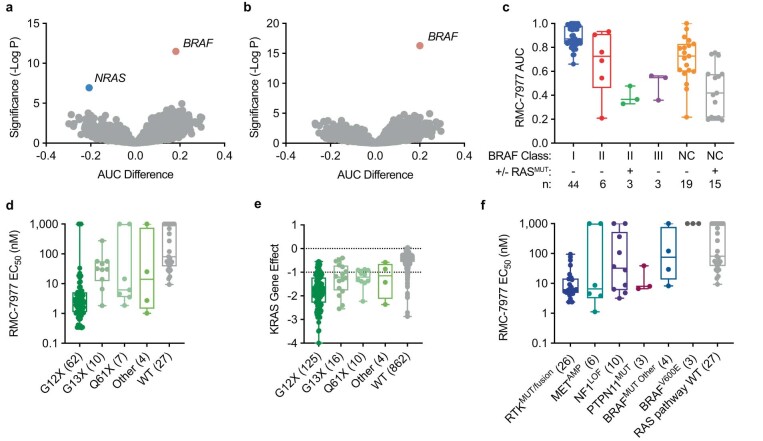
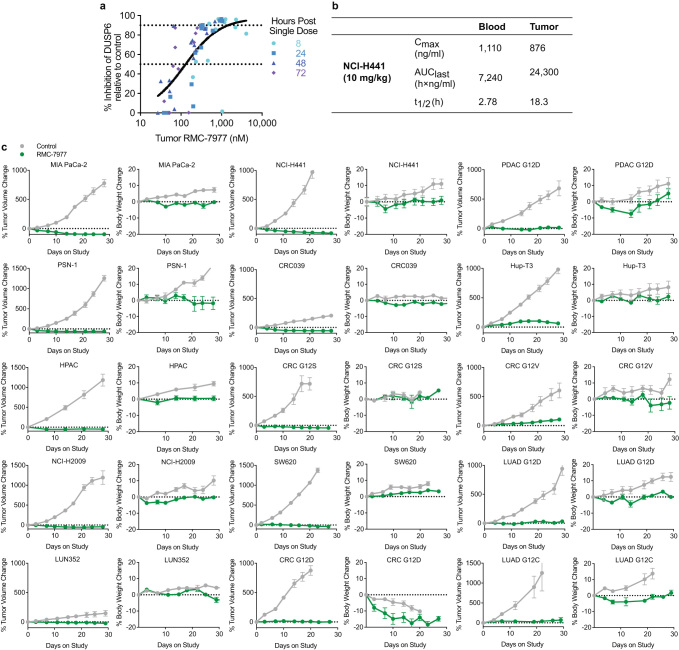
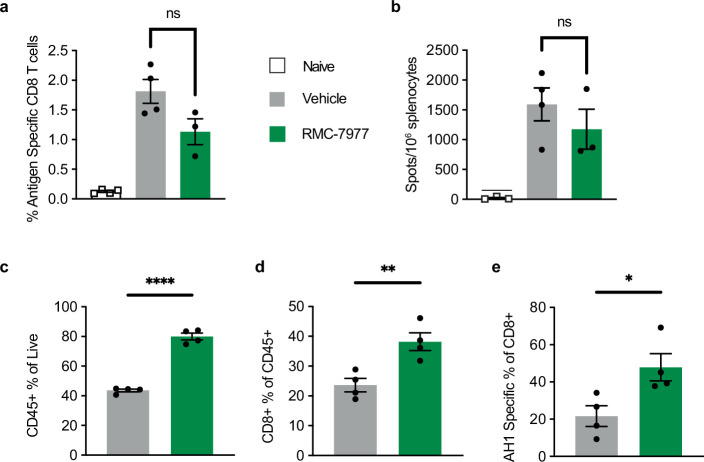
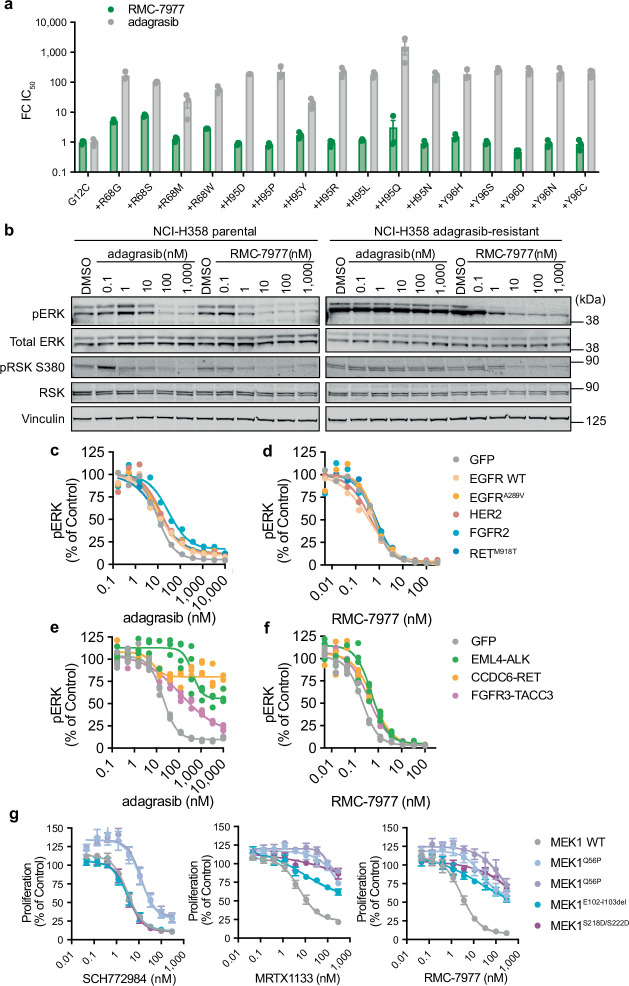
Comment in
-
New route to target RAS.Nat Rev Drug Discov. 2024 Jun;23(6):418. doi: 10.1038/d41573-024-00082-8. Nat Rev Drug Discov. 2024. PMID: 38730259 No abstract available.
References
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
