

Anoxia-induced hippocampal LTP is regeneratively produced by glutamate and nitric oxide from the neuro-glial-endothelial axis
- PMID: 38591010
- PMCID: PMC11000013
- DOI: 10.1016/j.isci.2024.109515
Anoxia-induced hippocampal LTP is regeneratively produced by glutamate and nitric oxide from the neuro-glial-endothelial axis
Abstract
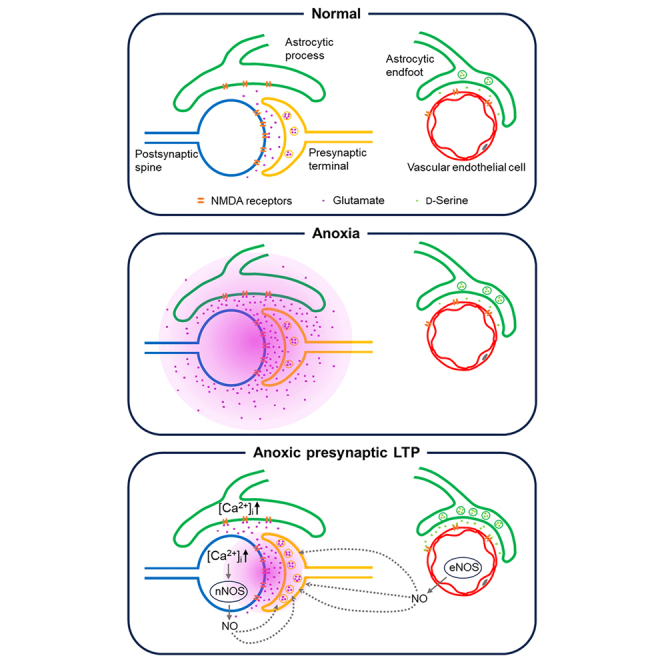
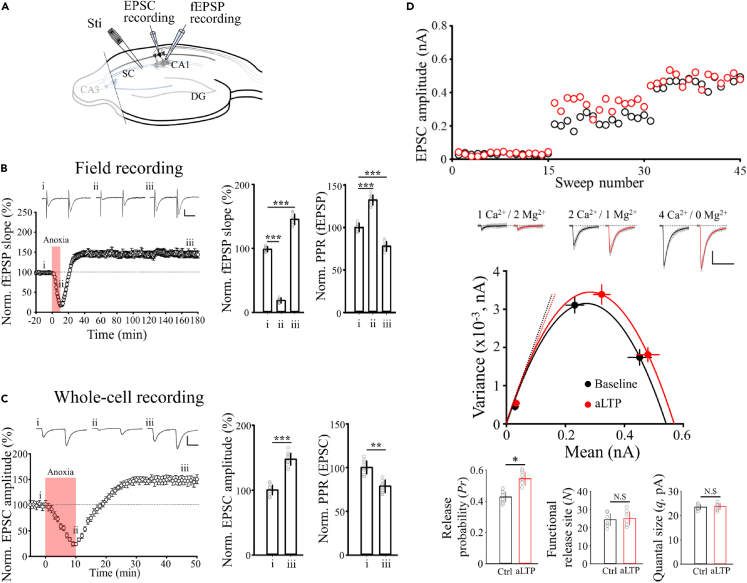
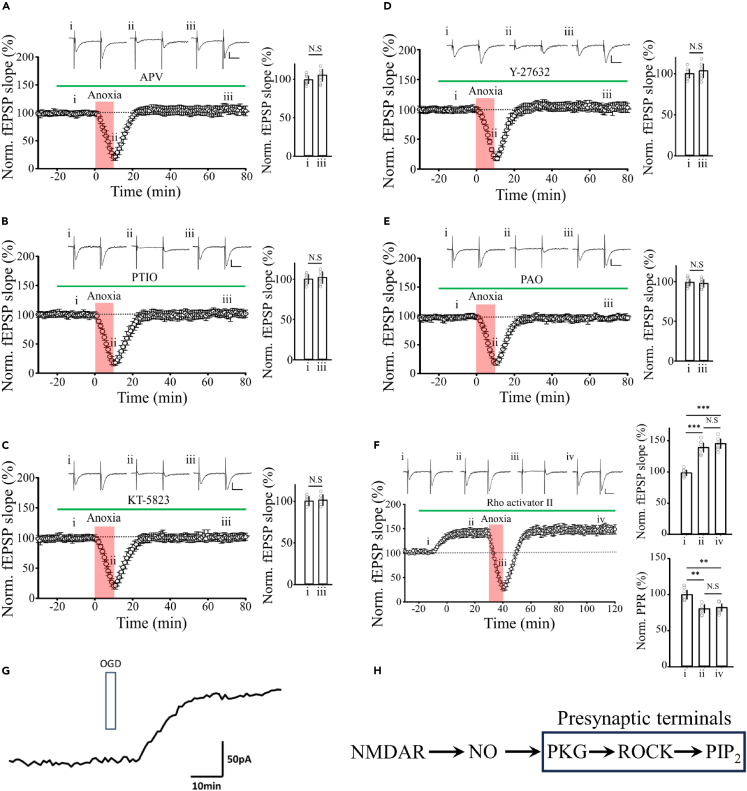
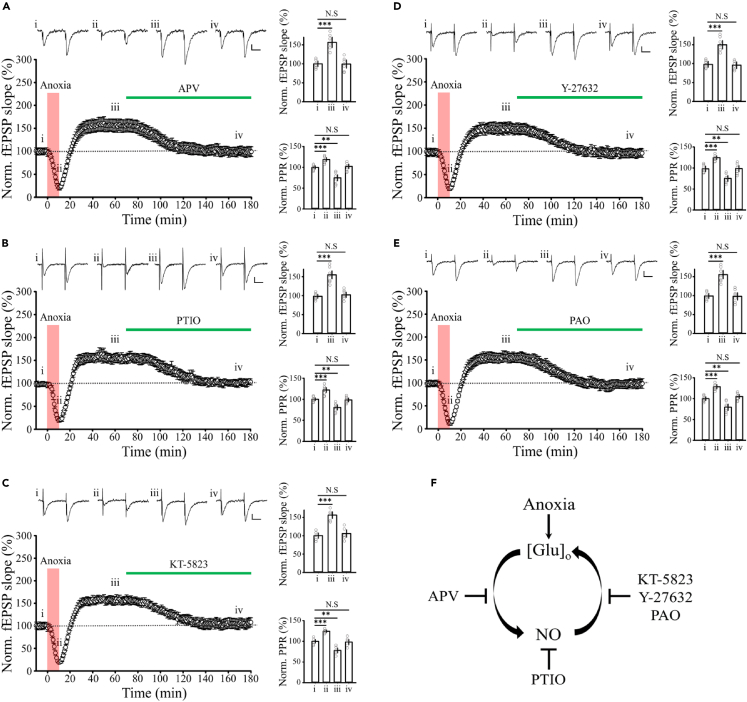
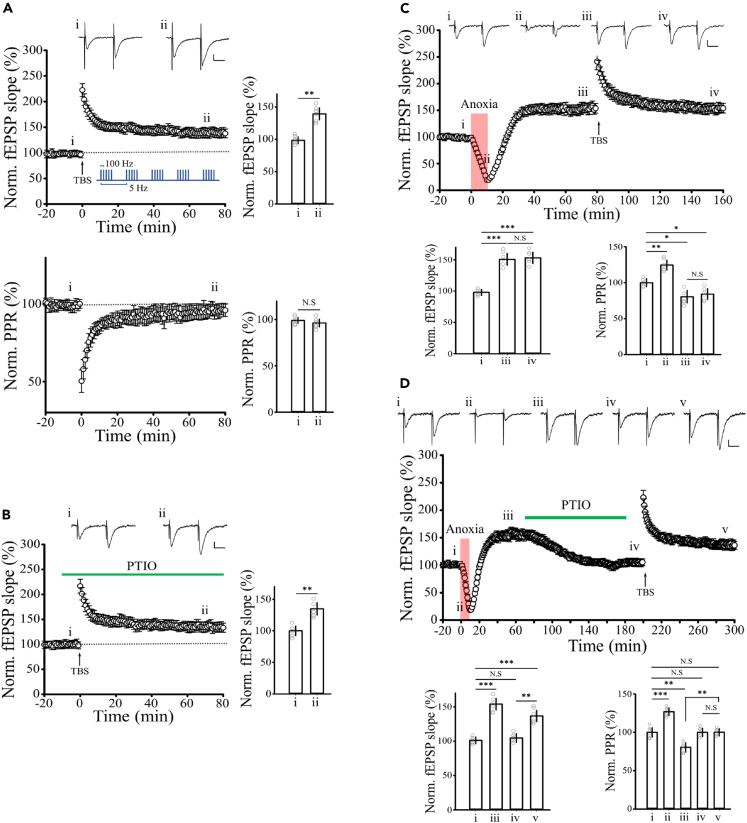
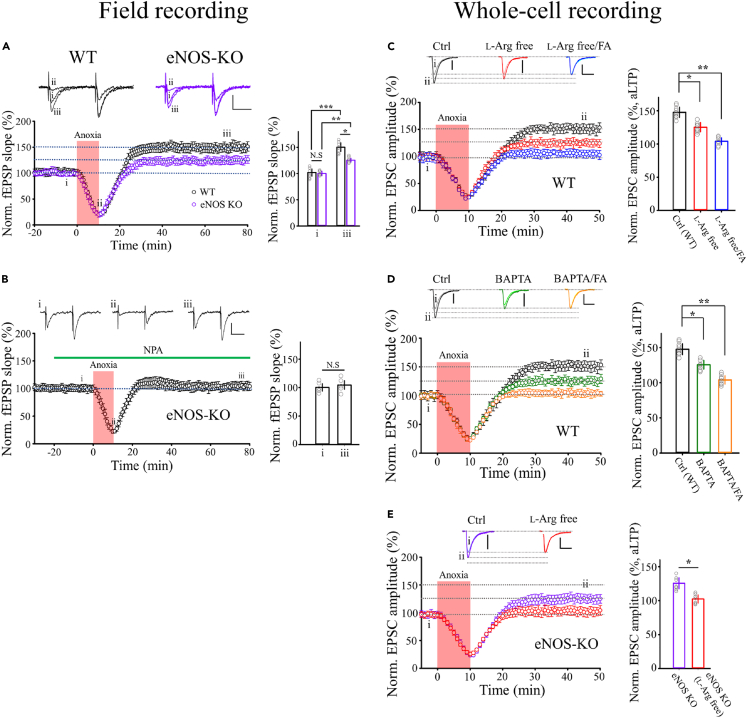
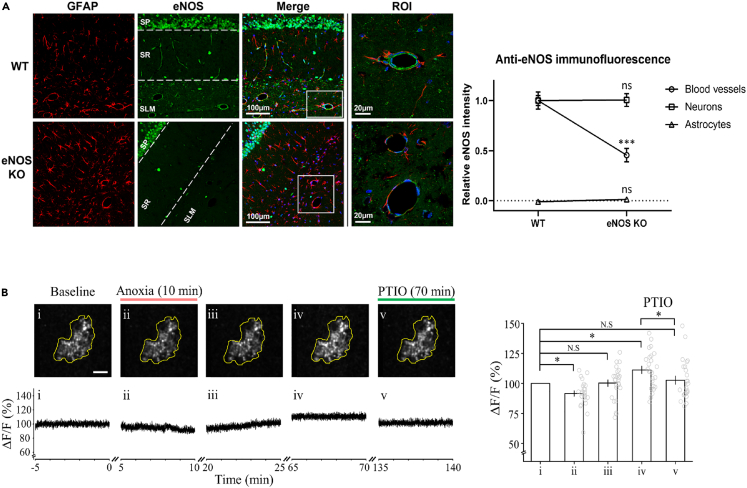
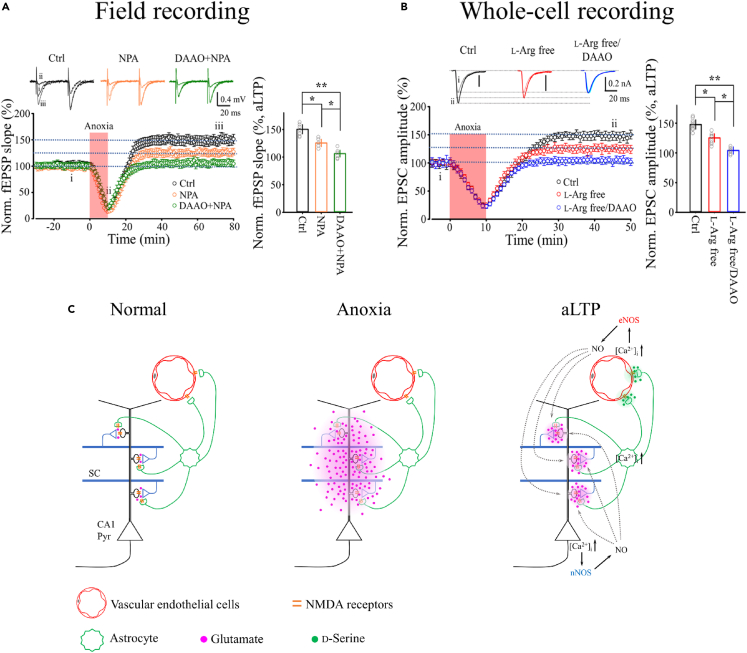
Transient anoxia causes amnesia and neuronal death. This is attributed to enhanced glutamate release and modeled as anoxia-induced long-term potentiation (aLTP). aLTP is mediated by glutamate receptors and nitric oxide (·NO) and occludes stimulation-induced LTP. We identified a signaling cascade downstream of ·NO leading to glutamate release and a glutamate-·NO loop regeneratively boosting aLTP. aLTP in entothelial ·NO synthase (eNOS)-knockout mice and blocking neuronal NOS (nNOS) activity suggested that both nNOS and eNOS contribute to aLTP. Immunostaining result showed that eNOS is predominantly expressed in vascular endothelia. Transient anoxia induced a long-lasting Ca2+ elevation in astrocytes that mirrored aLTP. Blocking astrocyte metabolism or depletion of the NMDA receptor ligand D-serine abolished eNOS-dependent aLTP, suggesting that astrocytic Ca2+ elevation stimulates D-serine release from endfeet to endothelia, thereby releasing ·NO synthesized by eNOS. Thus, the neuro-glial-endothelial axis is involved in long-term enhancement of glutamate release after transient anoxia.
Keywords: Biophysics; Molecular biology.
© 2024 The Authors.
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
Endothelial nitric oxide synthase is predominantly involved in angiotensin II modulation of renal vascular resistance and norepinephrine release.Am J Physiol Regul Integr Comp Physiol. 2008 Feb;294(2):R421-8. doi: 10.1152/ajpregu.00481.2007. Epub 2007 Nov 28. Am J Physiol Regul Integr Comp Physiol. 2008. PMID: 18046021
-
Control of the neurovascular coupling by nitric oxide-dependent regulation of astrocytic Ca(2+) signaling.Front Cell Neurosci. 2015 Mar 10;9:59. doi: 10.3389/fncel.2015.00059. eCollection 2015. Front Cell Neurosci. 2015. PMID: 25805969 Free PMC article. Review.
-
Neuronal nitric oxide synthase (NOS) regulates leukocyte-endothelial cell interactions in endothelial NOS deficient mice.Br J Pharmacol. 2001 Sep;134(2):305-12. doi: 10.1038/sj.bjp.0704234. Br J Pharmacol. 2001. PMID: 11564648 Free PMC article.
-
Endothelial and neuronal nitric oxide synthases are present in the suprachiasmatic nuclei of Syrian hamsters and rats.Eur J Neurosci. 2000 Feb;12(2):649-61. doi: 10.1046/j.1460-9568.2000.00961.x. Eur J Neurosci. 2000. PMID: 10712645
-
Neuronal and endothelial nitric oxide synthase gene knockout mice.Braz J Med Biol Res. 1999 Nov;32(11):1353-9. doi: 10.1590/s0100-879x1999001100005. Braz J Med Biol Res. 1999. PMID: 10559836 Review.
Cited by
-
The Unexpected Role of the Endothelial Nitric Oxide Synthase at the Neurovascular Unit: Beyond the Regulation of Cerebral Blood Flow.Int J Mol Sci. 2024 Aug 21;25(16):9071. doi: 10.3390/ijms25169071. Int J Mol Sci. 2024. PMID: 39201757 Free PMC article. Review.
-
Necroptosis in vascular cognitive impairment: mechanisms and therapeutic potential.Front Aging Neurosci. 2025 Jun 25;17:1599773. doi: 10.3389/fnagi.2025.1599773. eCollection 2025. Front Aging Neurosci. 2025. PMID: 40636901 Free PMC article. Review.
-
The Progress of Cognitive Dysfunction Impairment Caused by Temporal Lobe Epilepsy.J Mol Neurosci. 2025 Jun 21;75(3):81. doi: 10.1007/s12031-025-02365-0. J Mol Neurosci. 2025. PMID: 40542997 Free PMC article. Review.
-
Cognitive Impairment and Synaptic Dysfunction in Cardiovascular Disorders: The New Frontiers of the Heart-Brain Axis.Biomedicines. 2024 Oct 18;12(10):2387. doi: 10.3390/biomedicines12102387. Biomedicines. 2024. PMID: 39457698 Free PMC article. Review.
References
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
