

Targeting nucleotide metabolic pathways in colorectal cancer by integrating scRNA-seq, spatial transcriptome, and bulk RNA-seq data
- PMID: 38594466
- PMCID: PMC11004054
- DOI: 10.1007/s10142-024-01356-5
Targeting nucleotide metabolic pathways in colorectal cancer by integrating scRNA-seq, spatial transcriptome, and bulk RNA-seq data
Abstract
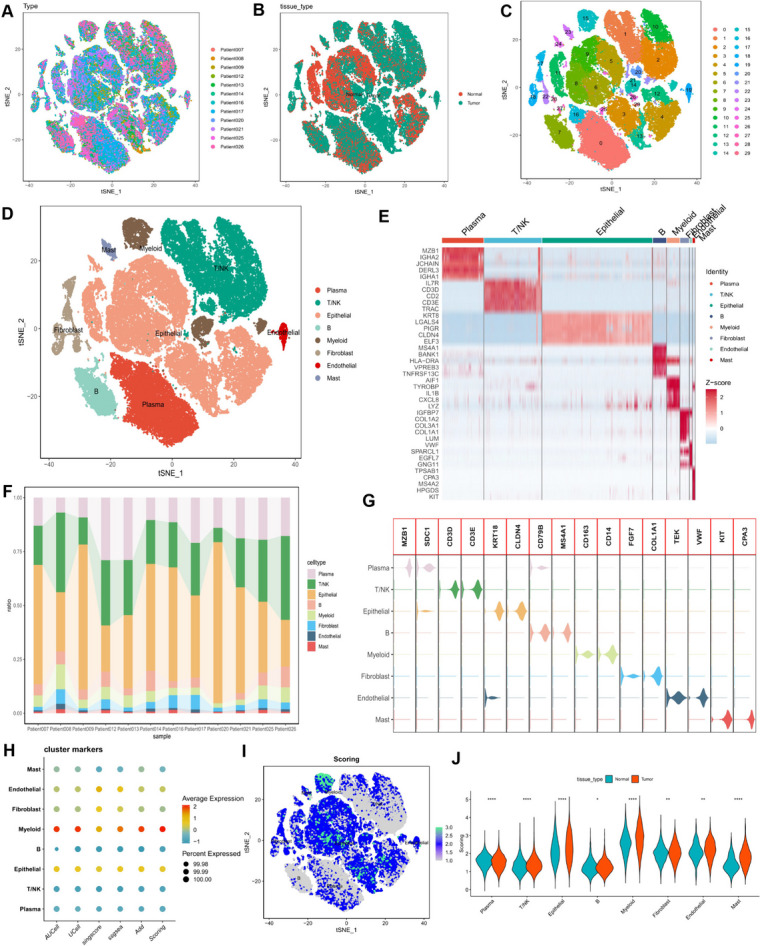
Background: Colorectal cancer is a malignant tumor of the digestive system originating from abnormal cell proliferation in the colon or rectum, often leading to gastrointestinal symptoms and severe health issues. Nucleotide metabolism, which encompasses the synthesis of DNA and RNA, is a pivotal cellular biochemical process that significantly impacts both the progression and therapeutic strategies of colorectal cancer METHODS: For single-cell RNA sequencing (scRNA-seq), five functions were employed to calculate scores related to nucleotide metabolism. Cell developmental trajectory analysis and intercellular interaction analysis were utilized to explore the metabolic characteristics and communication patterns of different epithelial cells. These findings were further validated using spatial transcriptome RNA sequencing (stRNA-seq). A risk model was constructed using expression profile data from TCGA and GEO cohorts to optimize clinical decision-making. Key nucleotide metabolism-related genes (NMRGs) were functionally validated by further in vitro experiments.
Results: In both scRNA-seq and stRNA-seq, colorectal cancer (CRC) exhibited unique cellular heterogeneity, with myeloid cells and epithelial cells in tumor samples displaying higher nucleotide metabolism scores. Analysis of intercellular communication revealed enhanced signaling pathways and ligand-receptor interactions between epithelial cells with high nucleotide metabolism and fibroblasts. Spatial transcriptome sequencing confirmed elevated nucleotide metabolism states in the core region of tumor tissue. After identifying differentially expressed NMRGs in epithelial cells, a risk prognostic model based on four genes effectively predicted overall survival and immunotherapy outcomes in patients. High-risk group patients exhibited an immunosuppressive microenvironment and relatively poorer prognosis and responses to chemotherapy and immunotherapy. Finally, based on data analysis and a series of cellular functional experiments, ACOX1 and CPT2 were identified as novel therapeutic targets for CRC.
Conclusion: In this study, a comprehensive analysis of NMRGs in CRC was conducted using a combination of single-cell sequencing, spatial transcriptome sequencing, and high-throughput data. The prognostic model constructed with NMRGs shows potential as a standalone prognostic marker for colorectal cancer patients and may significantly influence the development of personalized treatment approaches for CRC.
Keywords: Colorectal cancer; Immunotherapy; Nucleotide metabolism; Prognostic model; scRNA-seq; stRNA-seq.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no conflicts of interest.
Figures











References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
