

Watching a signaling protein function: What has been learned over four decades of time-resolved studies of photoactive yellow protein
- PMID: 38595979
- PMCID: PMC11003764
- DOI: 10.1063/4.0000241
Watching a signaling protein function: What has been learned over four decades of time-resolved studies of photoactive yellow protein
Abstract
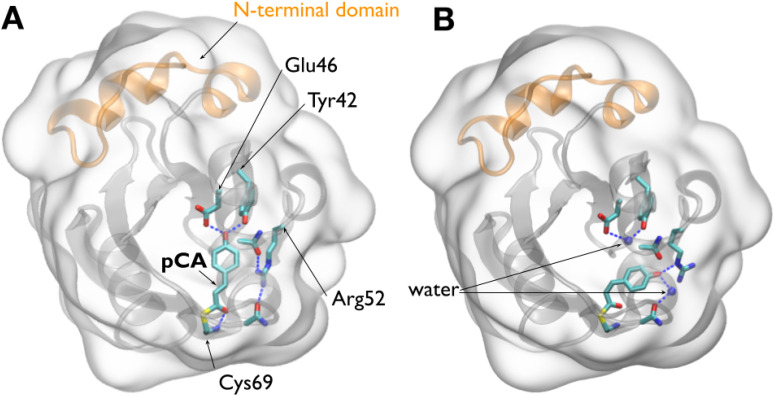
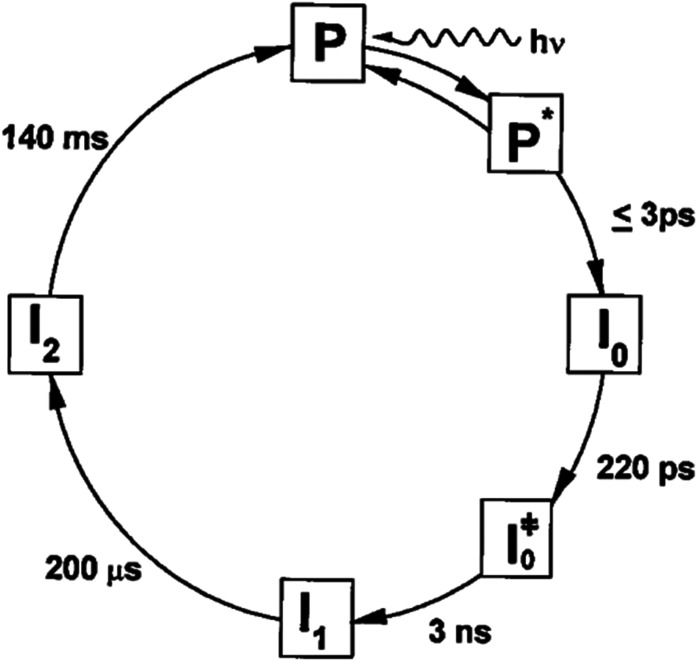
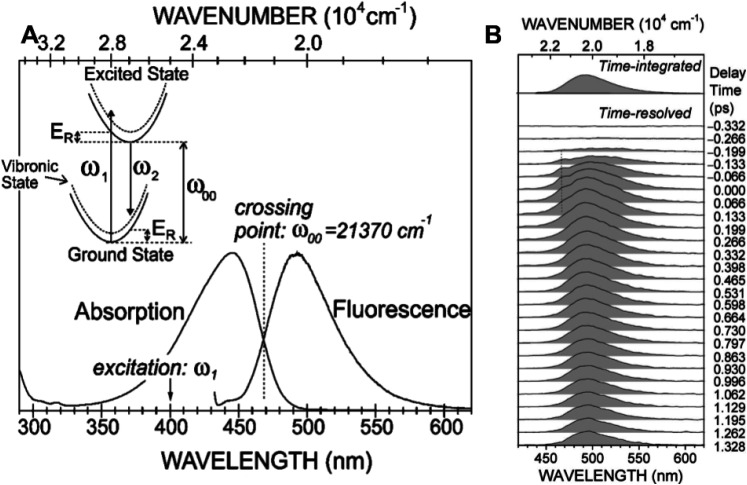
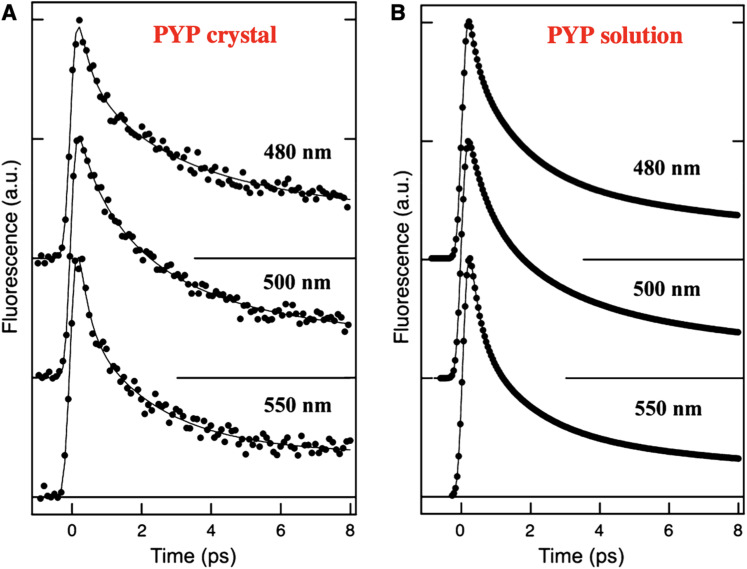
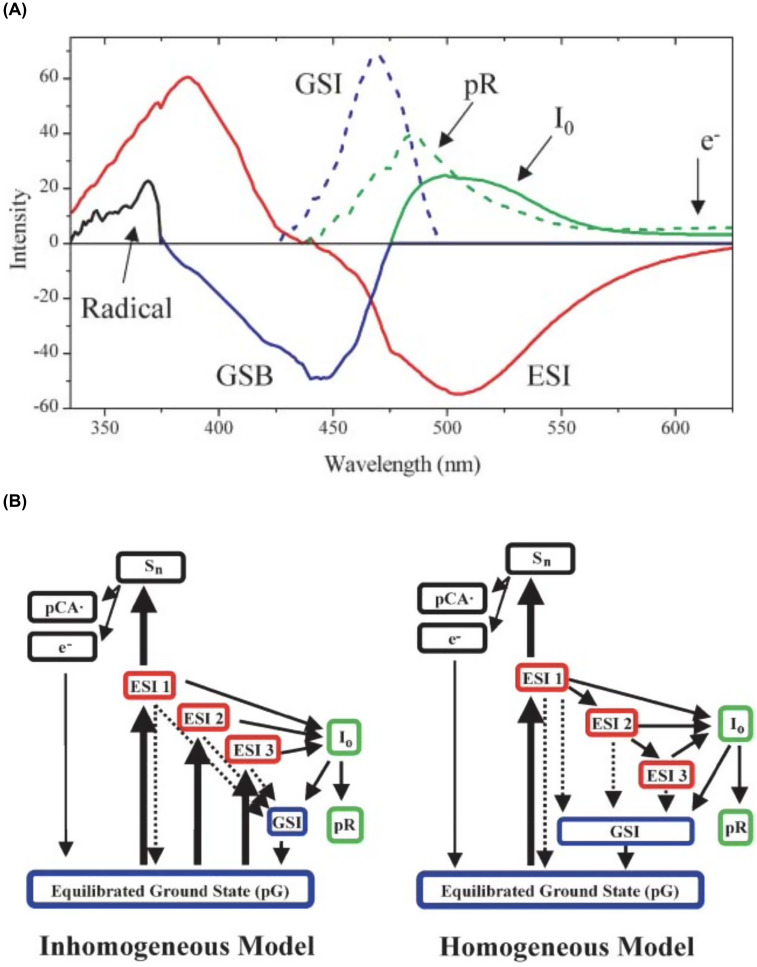
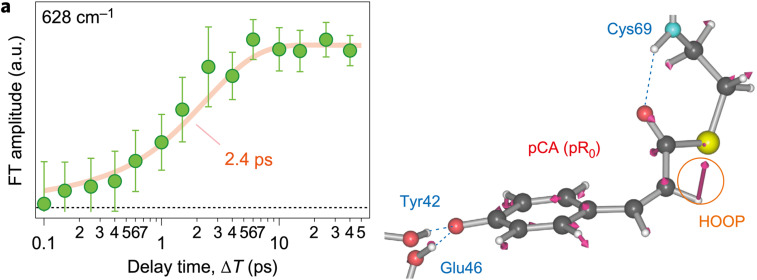
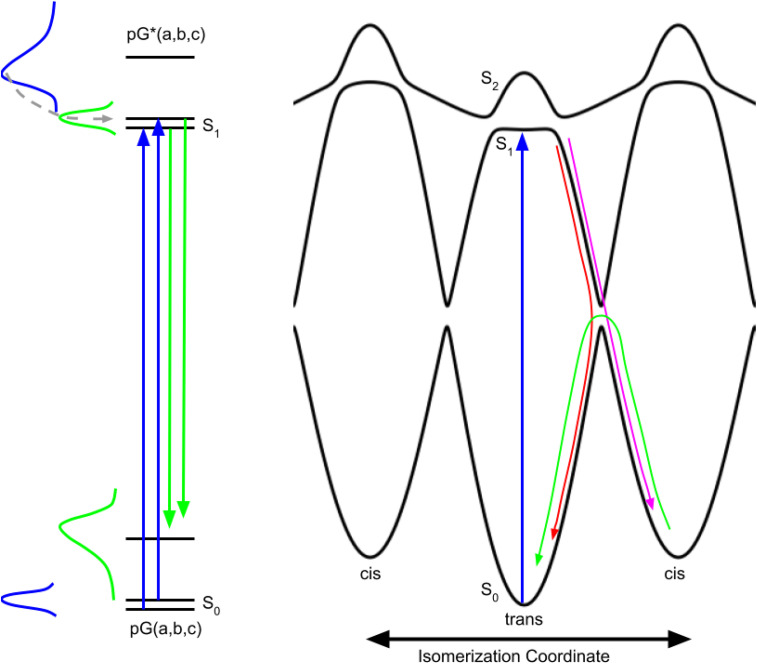
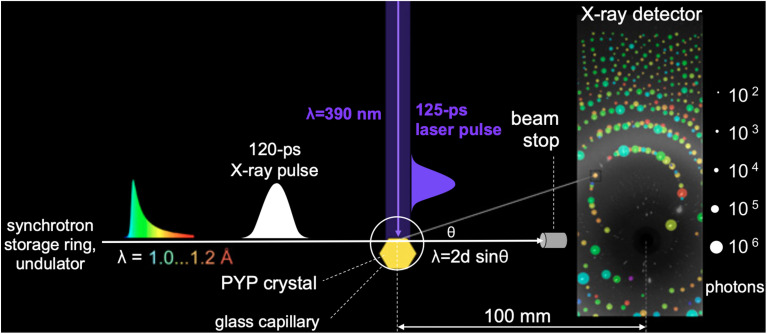
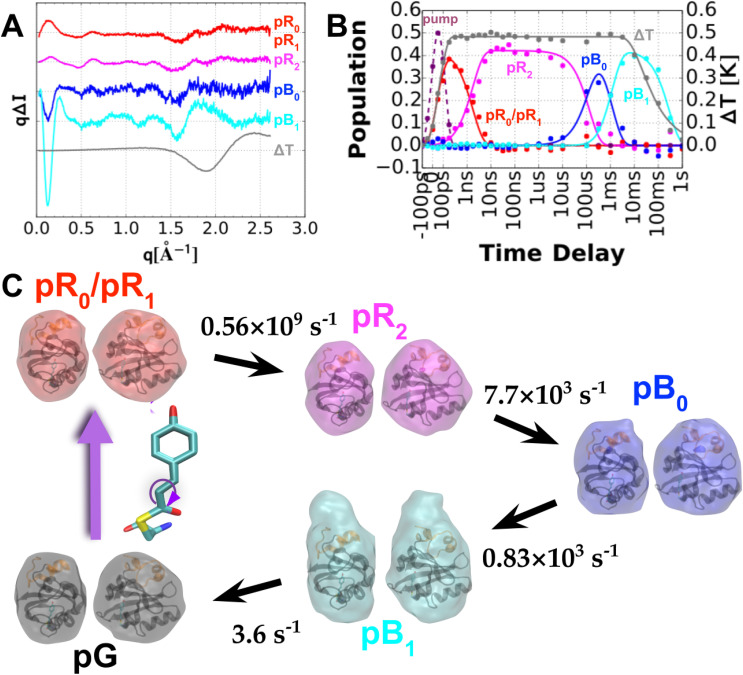
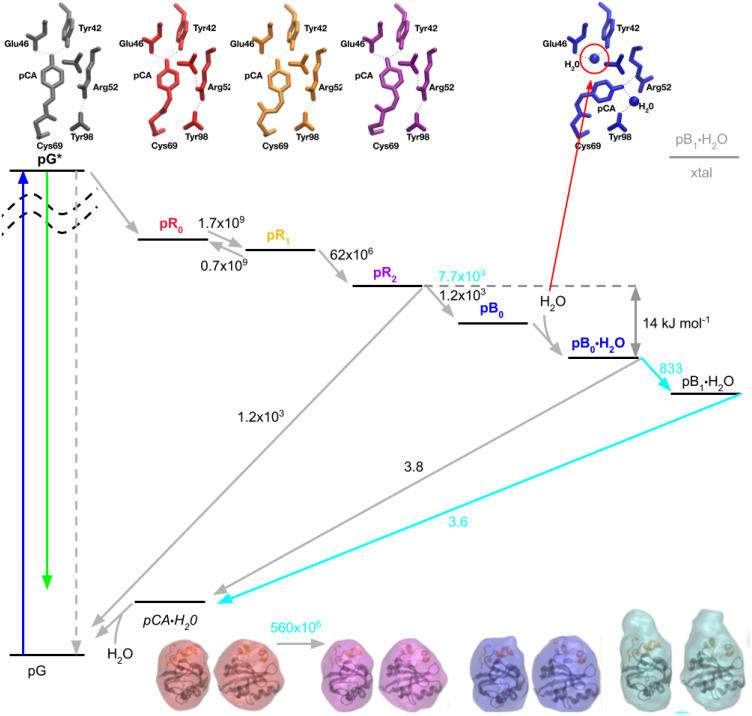
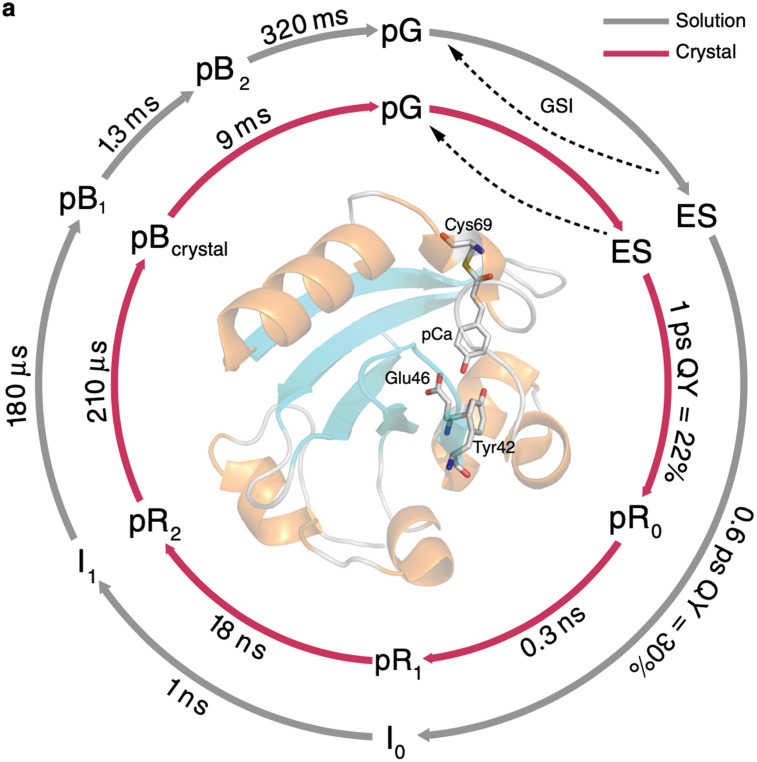
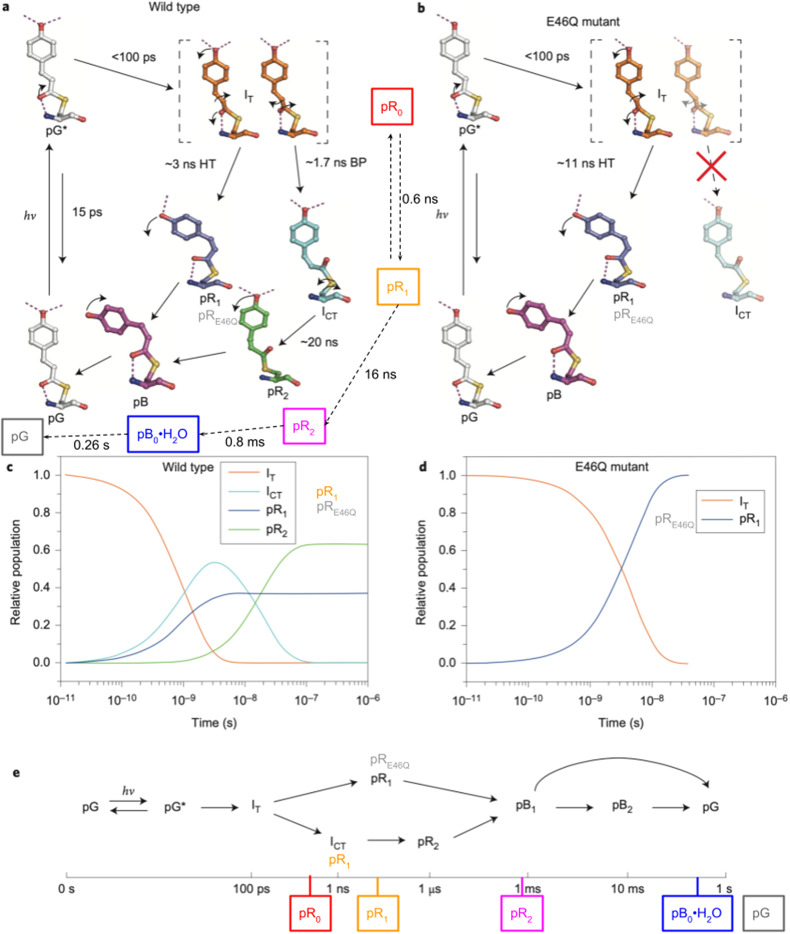
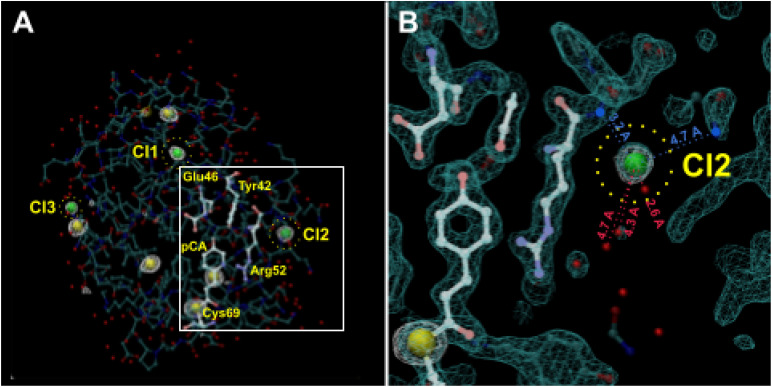
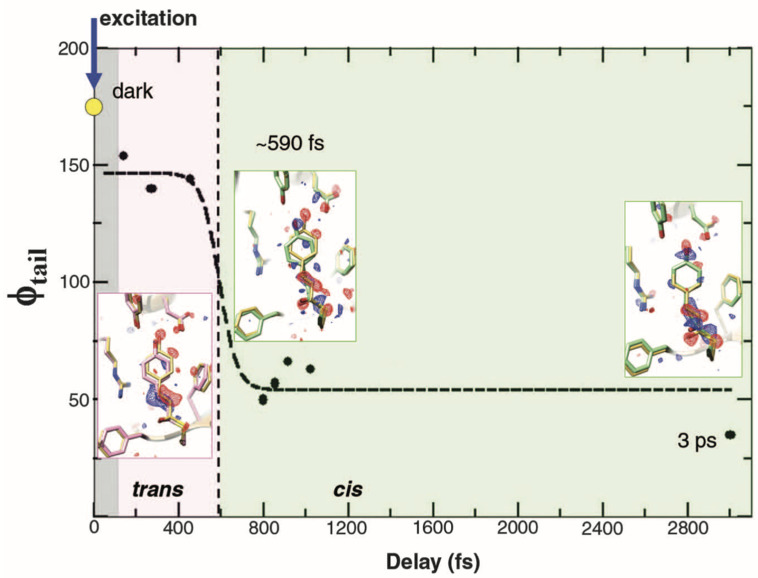
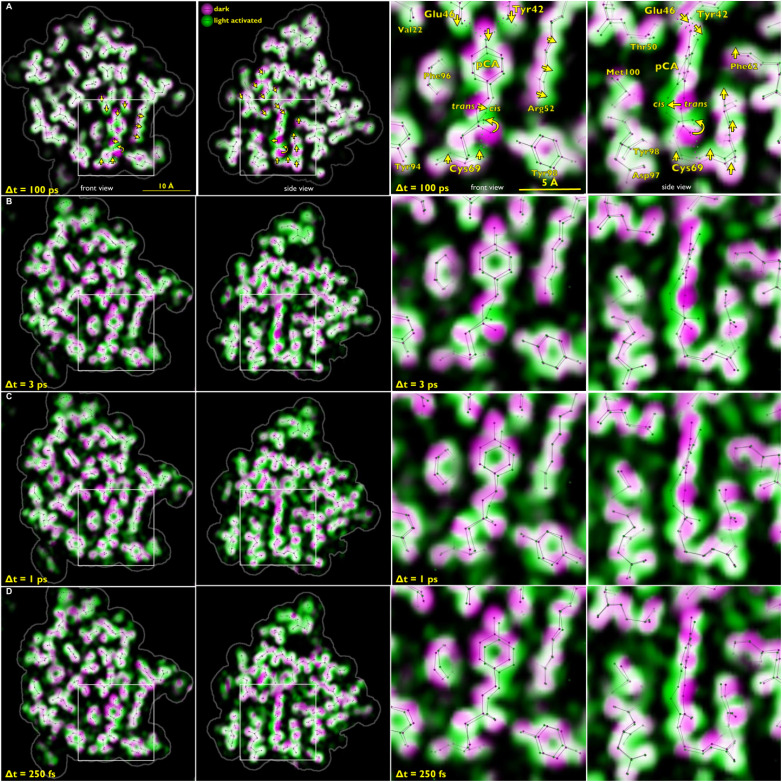
Photoactive yellow protein (PYP) is a signaling protein whose internal p-coumaric acid chromophore undergoes reversible, light-induced trans-to-cis isomerization, which triggers a sequence of structural changes that ultimately lead to a signaling state. Since its discovery nearly 40 years ago, PYP has attracted much interest and has become one of the most extensively studied proteins found in nature. The method of time-resolved crystallography, pioneered by Keith Moffat, has successfully characterized intermediates in the PYP photocycle at near atomic resolution over 12 decades of time down to the sub-picosecond time scale, allowing one to stitch together a movie and literally watch a protein as it functions. But how close to reality is this movie? To address this question, results from numerous complementary time-resolved techniques including x-ray crystallography, x-ray scattering, and spectroscopy are discussed. Emerging from spectroscopic studies is a general consensus that three time constants are required to model the excited state relaxation, with a highly strained ground-state cis intermediate formed in less than 2.4 ps. Persistent strain drives the sequence of structural transitions that ultimately produce the signaling state. Crystal packing forces produce a restoring force that slows somewhat the rates of interconversion between the intermediates. Moreover, the solvent composition surrounding PYP can influence the number and structures of intermediates as well as the rates at which they interconvert. When chloride is present, the PYP photocycle in a crystal closely tracks that in solution, which suggests the epic movie of the PYP photocycle is indeed based in reality.
© 2024 Author(s).
Conflict of interest statement
The authors have no conflicts to disclose.
Figures

Similar articles
-
Watching a signaling protein function in real time via 100-ps time-resolved Laue crystallography.Proc Natl Acad Sci U S A. 2012 Nov 20;109(47):19256-61. doi: 10.1073/pnas.1210938109. Epub 2012 Nov 6. Proc Natl Acad Sci U S A. 2012. PMID: 23132943 Free PMC article.
-
Molecular dynamics simulations of photoactive yellow protein (PYP) in three states of its photocycle: a comparison with X-ray and NMR data and analysis of the effects of Glu46 deprotonation and mutation.Eur Biophys J. 2002 Dec;31(7):504-20. doi: 10.1007/s00249-002-0243-1. Epub 2002 Oct 18. Eur Biophys J. 2002. PMID: 12451420
-
Picosecond Photobiology: Watching a Signaling Protein Function in Real Time via Time-Resolved Small- and Wide-Angle X-ray Scattering.J Am Chem Soc. 2016 Jul 20;138(28):8815-23. doi: 10.1021/jacs.6b03565. Epub 2016 Jul 5. J Am Chem Soc. 2016. PMID: 27305463 Free PMC article.
-
From primary photochemistry to biological function in the blue-light photoreceptors PYP and AppA.Photochem Photobiol Sci. 2005 Sep;4(9):688-93. doi: 10.1039/b418442b. Epub 2005 Apr 6. Photochem Photobiol Sci. 2005. PMID: 16121278 Review.
-
Initial photoinduced dynamics of the photoactive yellow protein.Chemphyschem. 2005 May;6(5):828-37. doi: 10.1002/cphc.200400351. Chemphyschem. 2005. PMID: 15884065 Review.
Cited by
-
Preface to special topic: Celebrating the work and achievements of Keith Moffat.Struct Dyn. 2025 Jul 1;12(4):040401. doi: 10.1063/4.0000771. eCollection 2025 Jul. Struct Dyn. 2025. PMID: 40612268 Free PMC article. No abstract available.
References
-
- Baca, M. , Borgstahl, G. E. , Boissinot, M. , Burke, P. M. , Williams, D. R. , Slater, K. A. , and Getzoff, E. D. , “ Complete chemical structure of photoactive yellow protein: Novel thioester-linked 4-hydroxycinnamyl chromophore and photocycle chemistry,” Biochemistry 33(48), 14369–14377 (1994).10.1021/bi00252a001 - DOI - PubMed
-
- Cho, H. S. , Schotte, F. , Dashdorj, N. , Kyndt, J. , Henning, R. , and Anfinrud, P. A. , “ Picosecond photobiology: Watching a signaling protein function in real time via time-resolved small- and wide-angle x-ray scattering,” J. Am. Chem. Soc. 138(28), 8815–8823 (2016).10.1021/jacs.6b03565 - DOI - PMC - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous