

Discovery and preclinical development of a therapeutically active nanobody-based chimeric antigen receptor targeting human CD22
- PMID: 38596311
- PMCID: PMC10914482
- DOI: 10.1016/j.omton.2024.200775
Discovery and preclinical development of a therapeutically active nanobody-based chimeric antigen receptor targeting human CD22
Abstract
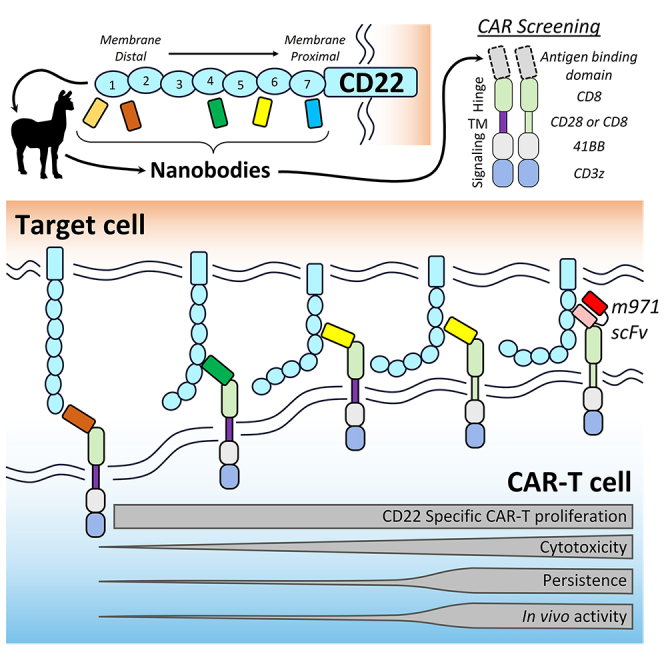
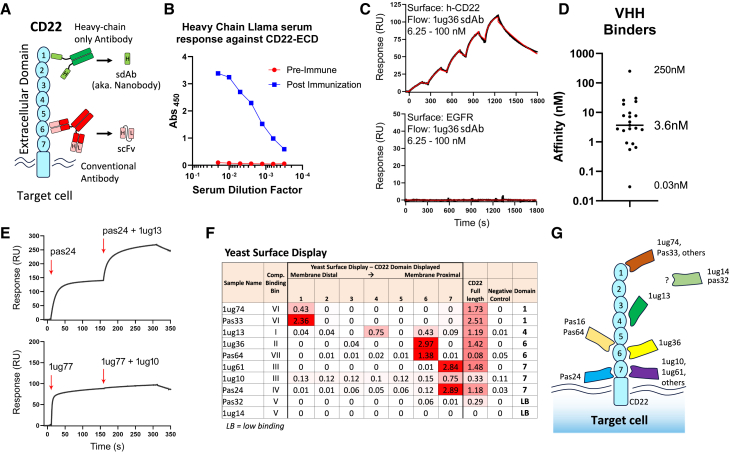
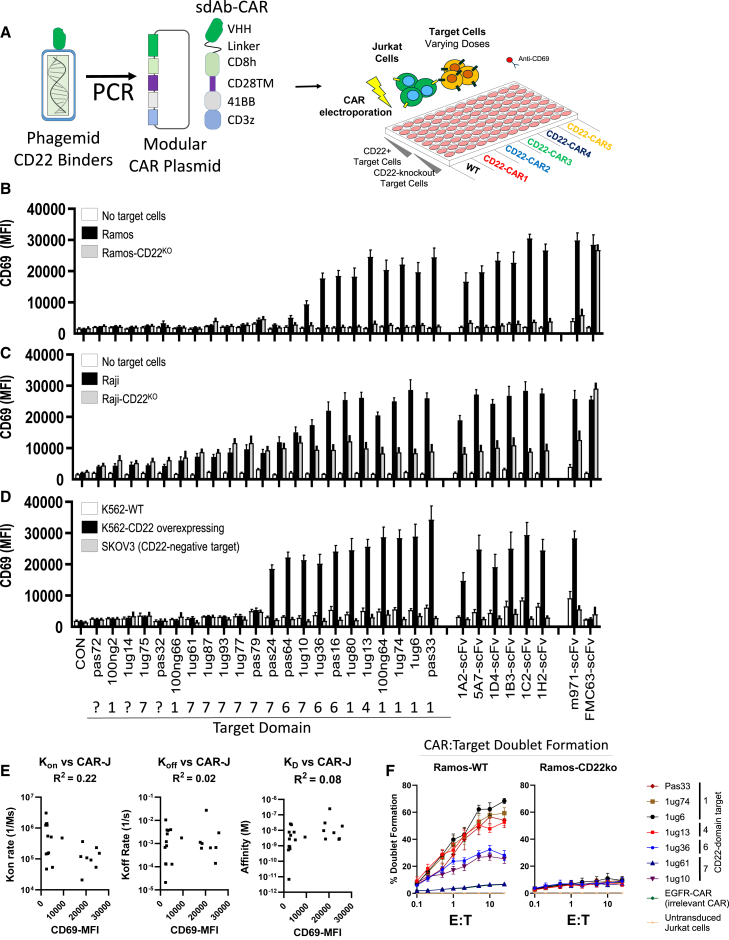
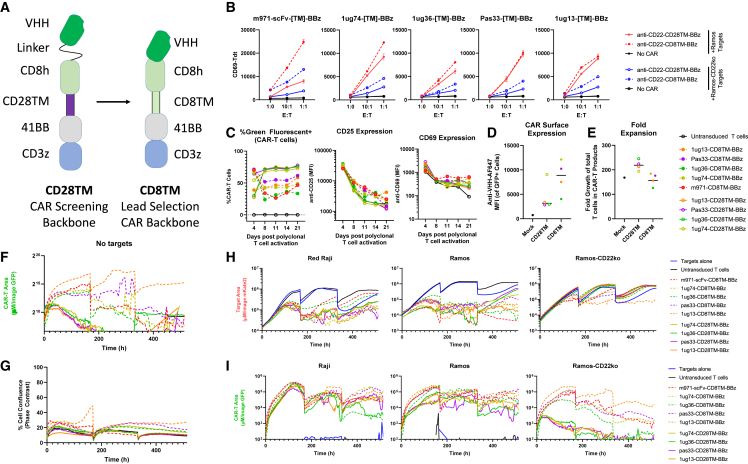
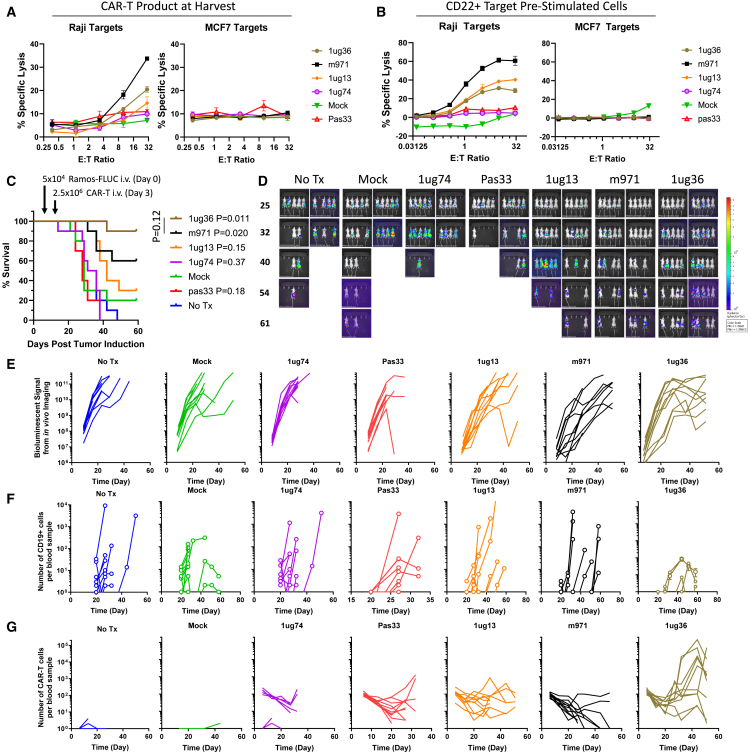
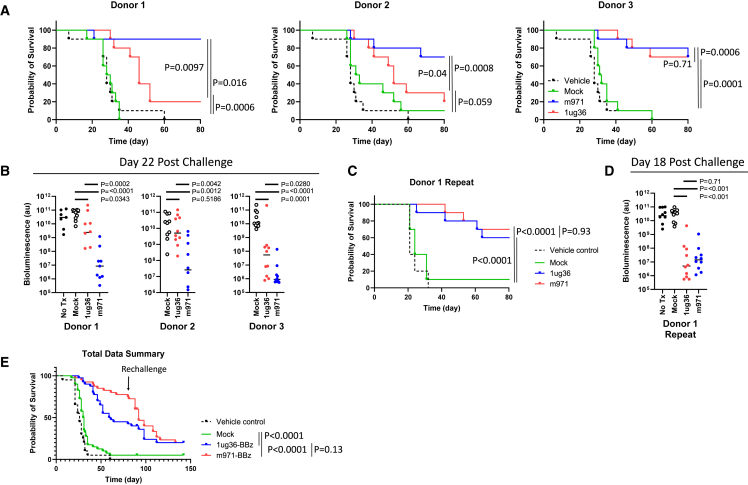
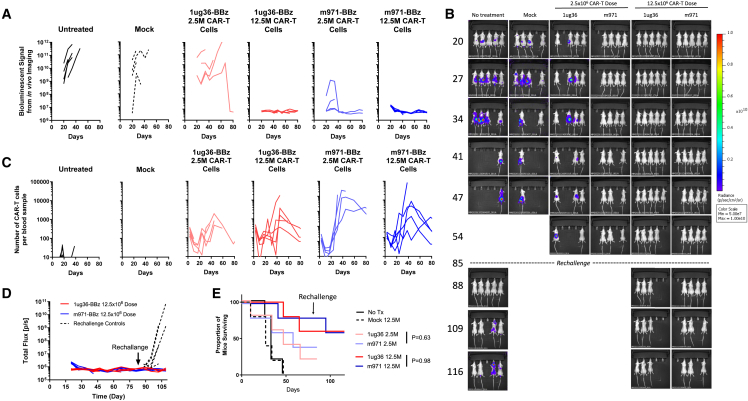
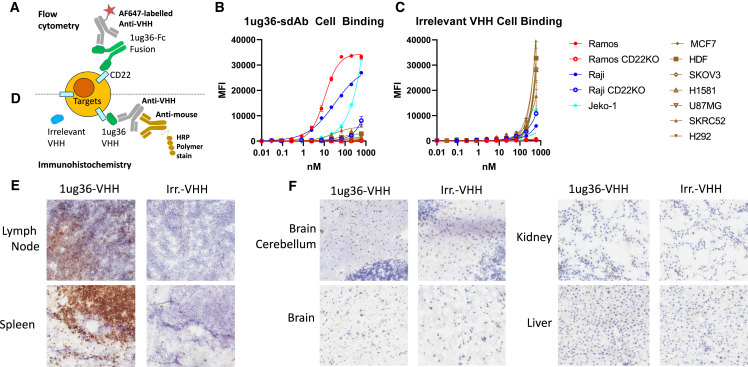
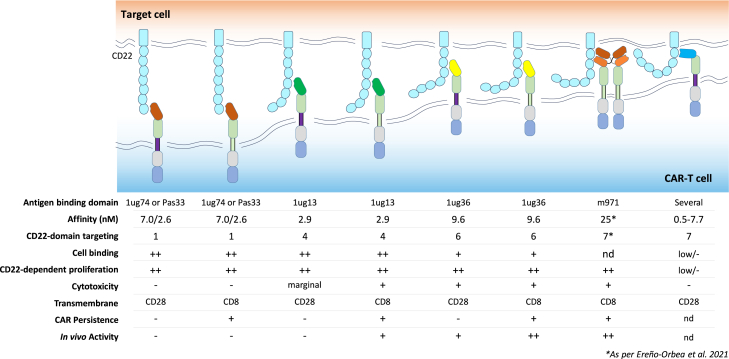
Chimeric antigen receptor (CAR) T cell therapies targeting B cell-restricted antigens CD19, CD20, or CD22 can produce potent clinical responses for some B cell malignancies, but relapse remains common. Camelid single-domain antibodies (sdAbs or nanobodies) are smaller, simpler, and easier to recombine than single-chain variable fragments (scFvs) used in most CARs, but fewer sdAb-CARs have been reported. Thus, we sought to identify a therapeutically active sdAb-CAR targeting human CD22. Immunization of an adult Llama glama with CD22 protein, sdAb-cDNA library construction, and phage panning yielded >20 sdAbs with diverse epitope and binding properties. Expressing CD22-sdAb-CAR in Jurkat cells drove varying CD22-specific reactivity not correlated with antibody affinity. Changing CD28- to CD8-transmembrane design increased CAR persistence and expression in vitro. CD22-sdAb-CAR candidates showed similar CD22-dependent CAR-T expansion in vitro, although only membrane-proximal epitope targeting CD22-sdAb-CARs activated direct cytolytic killing and extended survival in a lymphoma xenograft model. Based on enhanced survival in blinded xenograft studies, a lead CD22sdCAR-T was selected, achieving comparable complete responses to a benchmark short linker m971-scFv CAR-T in high-dose experiments. Finally, immunohistochemistry and flow cytometry confirm tissue and cellular-level specificity of the lead CD22-sdAb. This presents a complete report on preclinical development of a novel CD22sdCAR therapeutic.
Keywords: CAR optimization; CAR-T; CD22; MT: Regular Issue; cell therapy; chimeric antigen receptors; hematological malignancy; leukemia and lymphoma; nanobody; preclinical development; single-domain antibody.
Crown Copyright © 2024.
Conflict of interest statement
CD22-nanobody binding elements used in this work were disclosed in provisional patent filing US 2023/0265185 A1 and other provisional filings by the NRC. The authors declare no competing interests.
Figures
References
LinkOut - more resources
Full Text Sources
Research Materials