

Interstitial macrophages are a focus of viral takeover and inflammation in COVID-19 initiation in human lung
- PMID: 38597954
- PMCID: PMC11009983
- DOI: 10.1084/jem.20232192
Interstitial macrophages are a focus of viral takeover and inflammation in COVID-19 initiation in human lung
Abstract
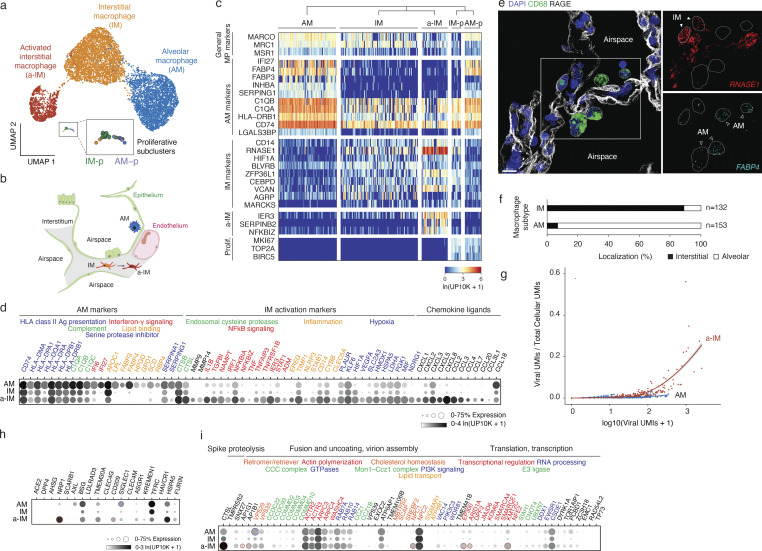
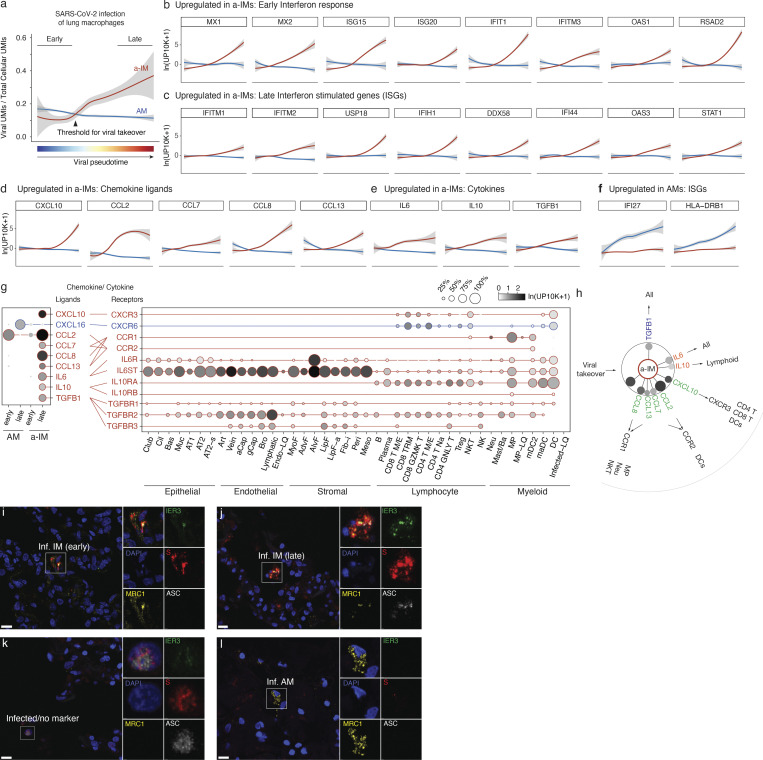
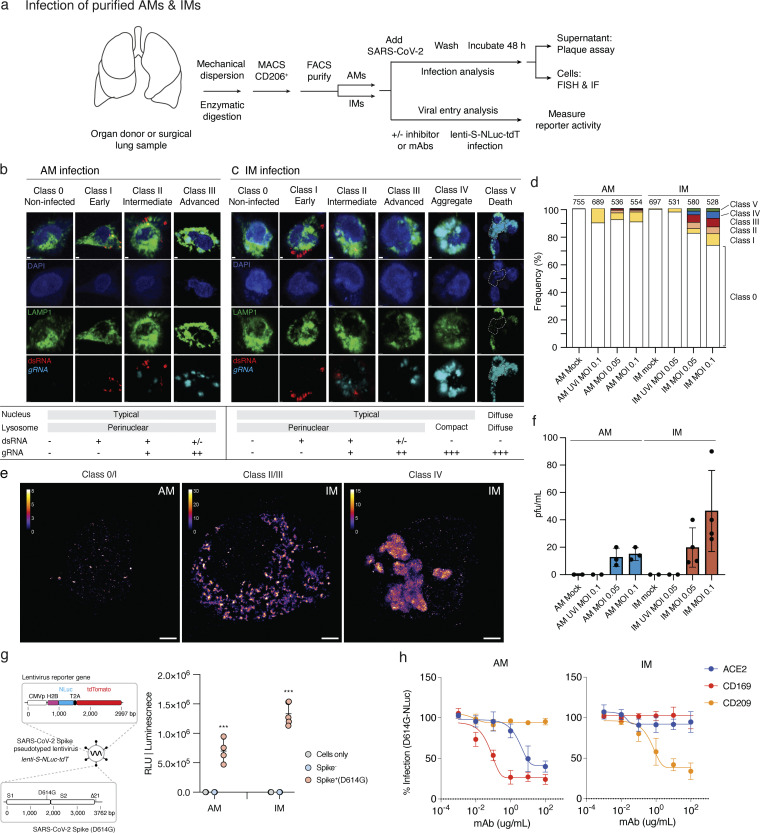
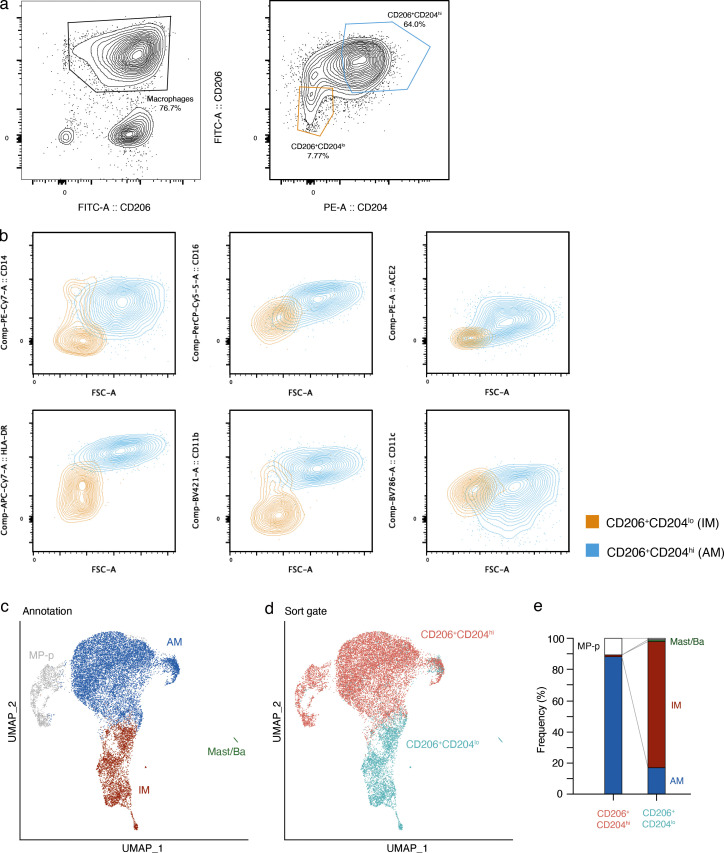
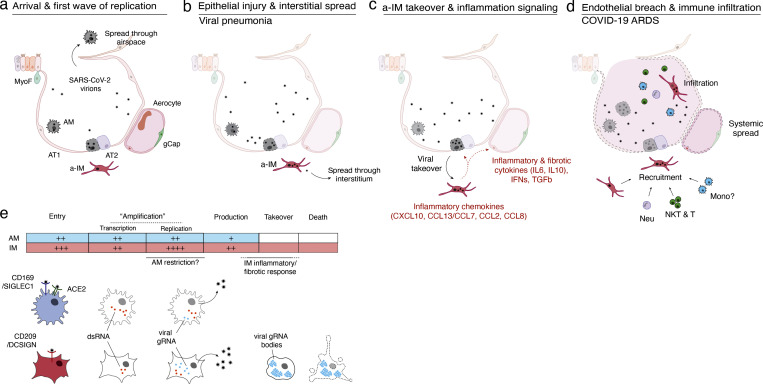
Early stages of deadly respiratory diseases including COVID-19 are challenging to elucidate in humans. Here, we define cellular tropism and transcriptomic effects of SARS-CoV-2 virus by productively infecting healthy human lung tissue and using scRNA-seq to reconstruct the transcriptional program in "infection pseudotime" for individual lung cell types. SARS-CoV-2 predominantly infected activated interstitial macrophages (IMs), which can accumulate thousands of viral RNA molecules, taking over 60% of the cell transcriptome and forming dense viral RNA bodies while inducing host profibrotic (TGFB1, SPP1) and inflammatory (early interferon response, CCL2/7/8/13, CXCL10, and IL6/10) programs and destroying host cell architecture. Infected alveolar macrophages (AMs) showed none of these extreme responses. Spike-dependent viral entry into AMs used ACE2 and Sialoadhesin/CD169, whereas IM entry used DC-SIGN/CD209. These results identify activated IMs as a prominent site of viral takeover, the focus of inflammation and fibrosis, and suggest targeting CD209 to prevent early pathology in COVID-19 pneumonia. This approach can be generalized to any human lung infection and to evaluate therapeutics.
© 2024 Wu et al.
Conflict of interest statement
Disclosures: J.B. Shrager reported personal fees from Merck, Inc. outside the submitted work. No other disclosures were reported.
Figures
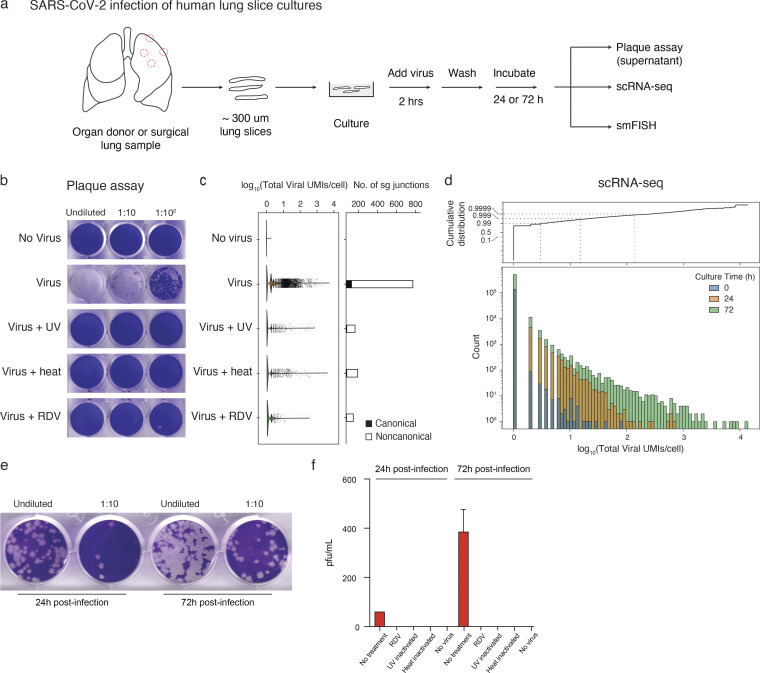
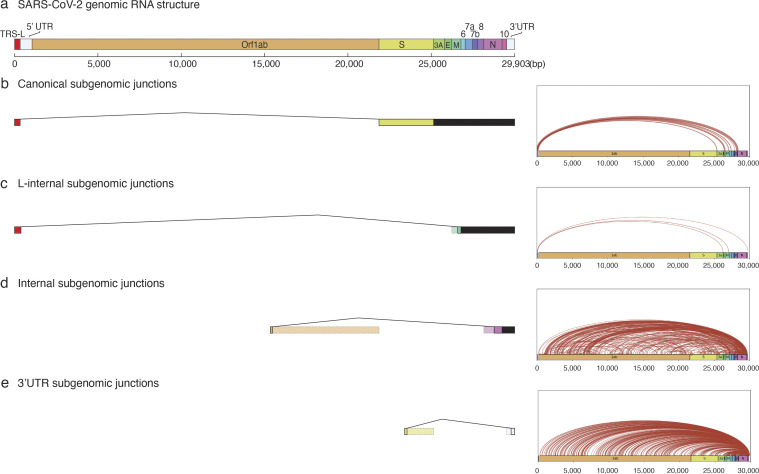
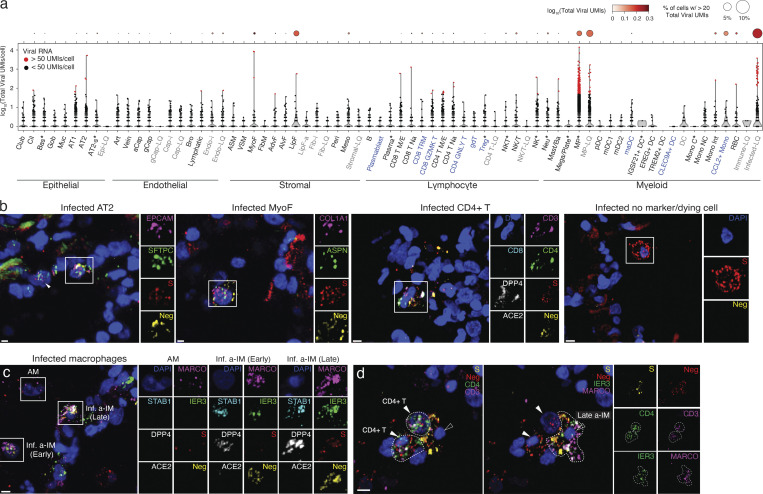
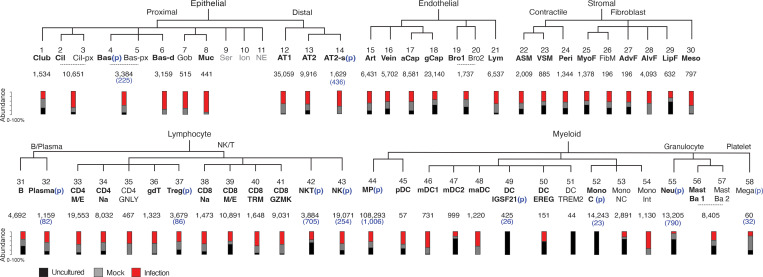


References
-
- Adams, T.S., Schupp J.C., Poli S., Ayaub E.A., Neumark N., Ahangari F., Chu S.G., Raby B.A., DeIuliis G., Januszyk M., et al. . 2020. Single-cell RNA-seq reveals ectopic and aberrant lung-resident cell populations in idiopathic pulmonary fibrosis. Sci. Adv. 6:eaba1983. 10.1126/sciadv.aba1983 - DOI - PMC - PubMed
-
- Anderson, R.N.; CDC . 1999. Deaths: Leading causes for 1999. Natl. Vital Stat. Rep. 49:1–87. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
