

Potentiating Activity of GmhA Inhibitors on Gram-Negative Bacteria
- PMID: 38598312
- PMCID: PMC11056994
- DOI: 10.1021/acs.jmedchem.4c00037
Potentiating Activity of GmhA Inhibitors on Gram-Negative Bacteria
Abstract
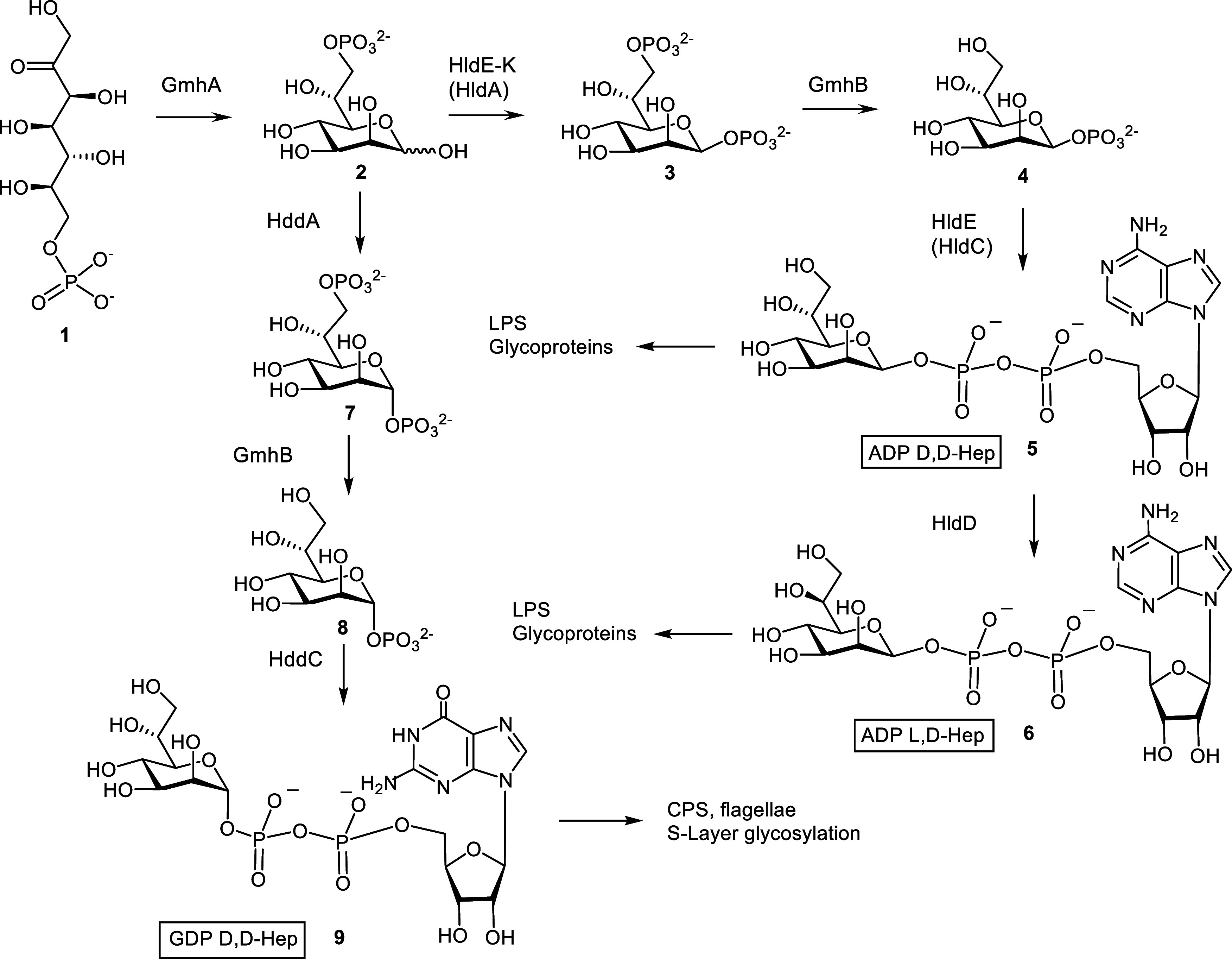
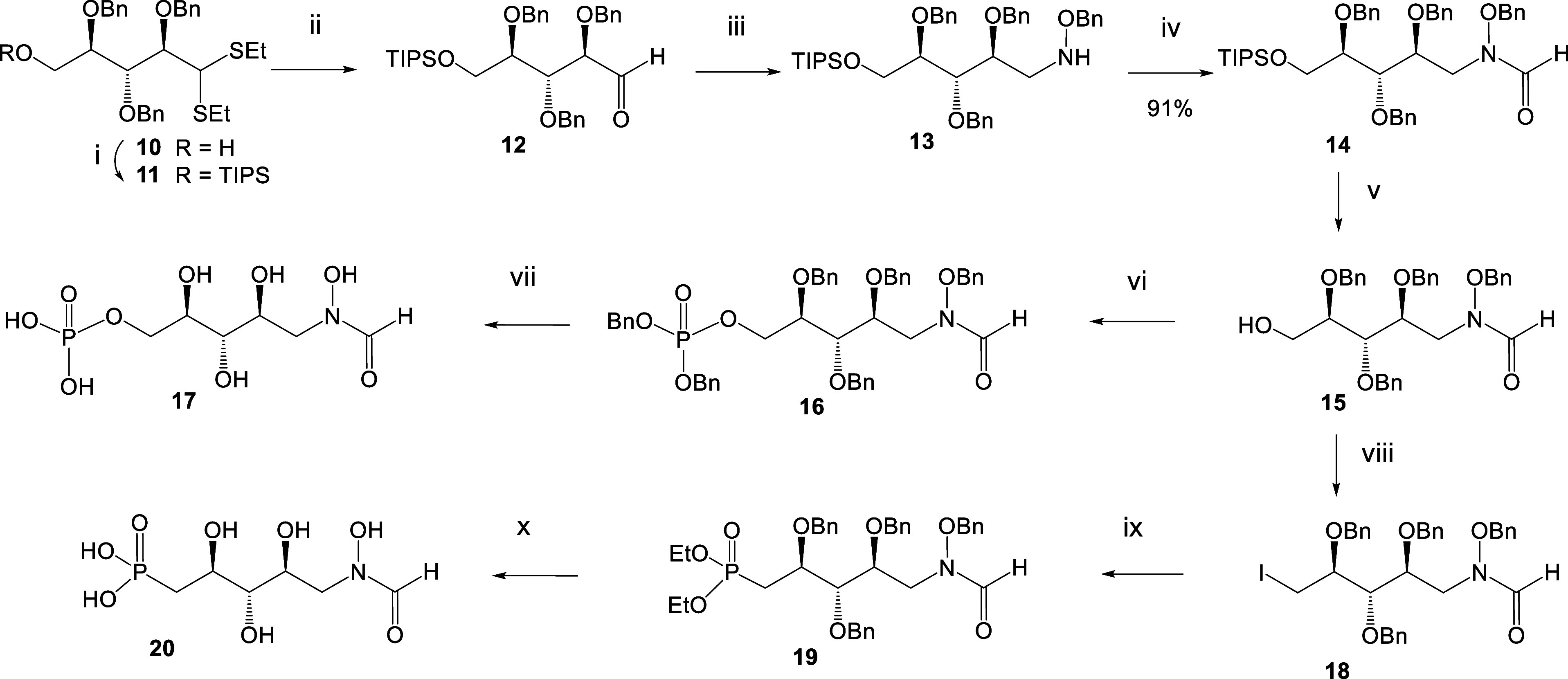
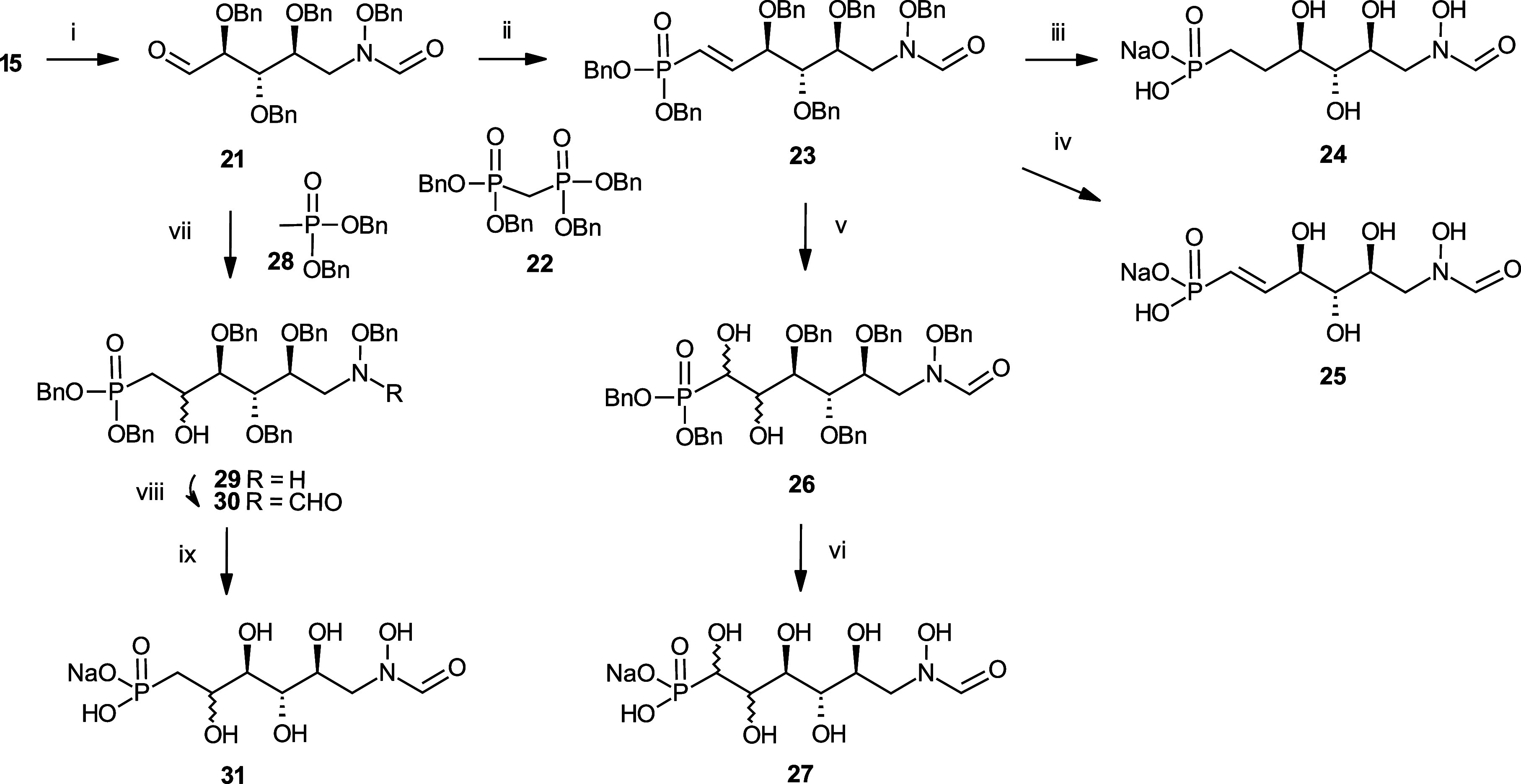
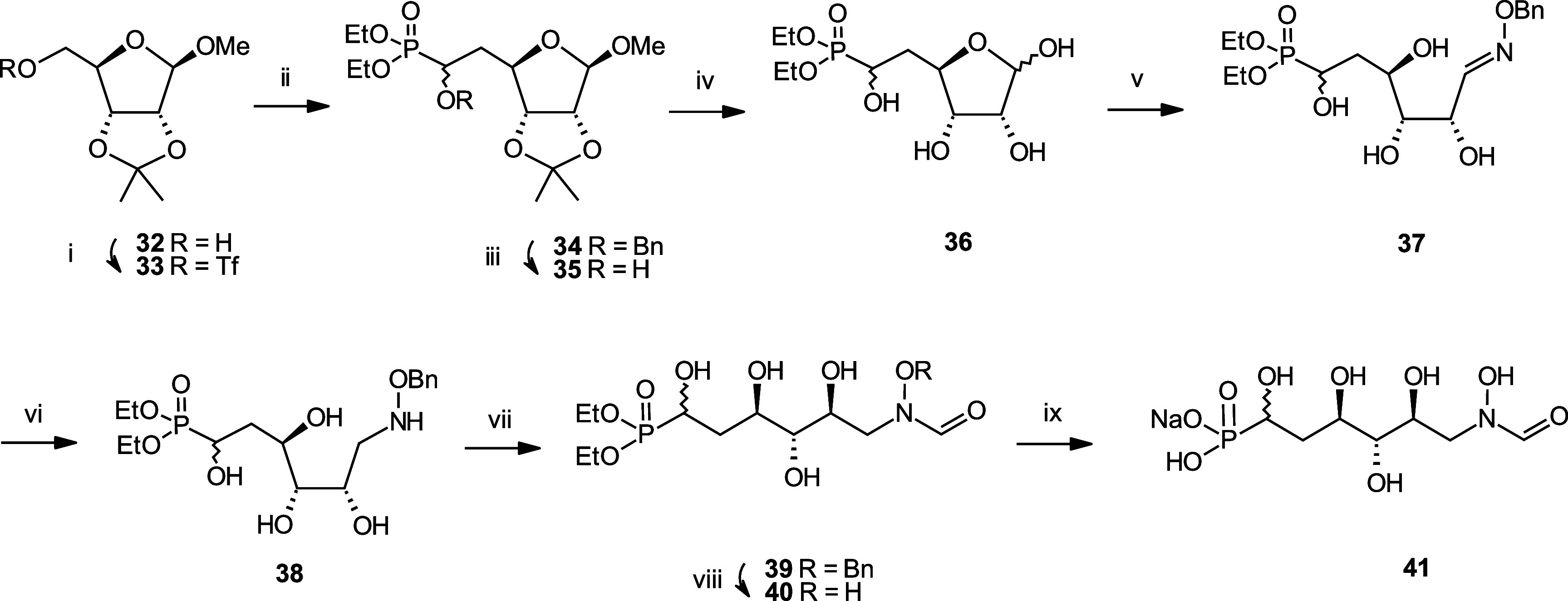
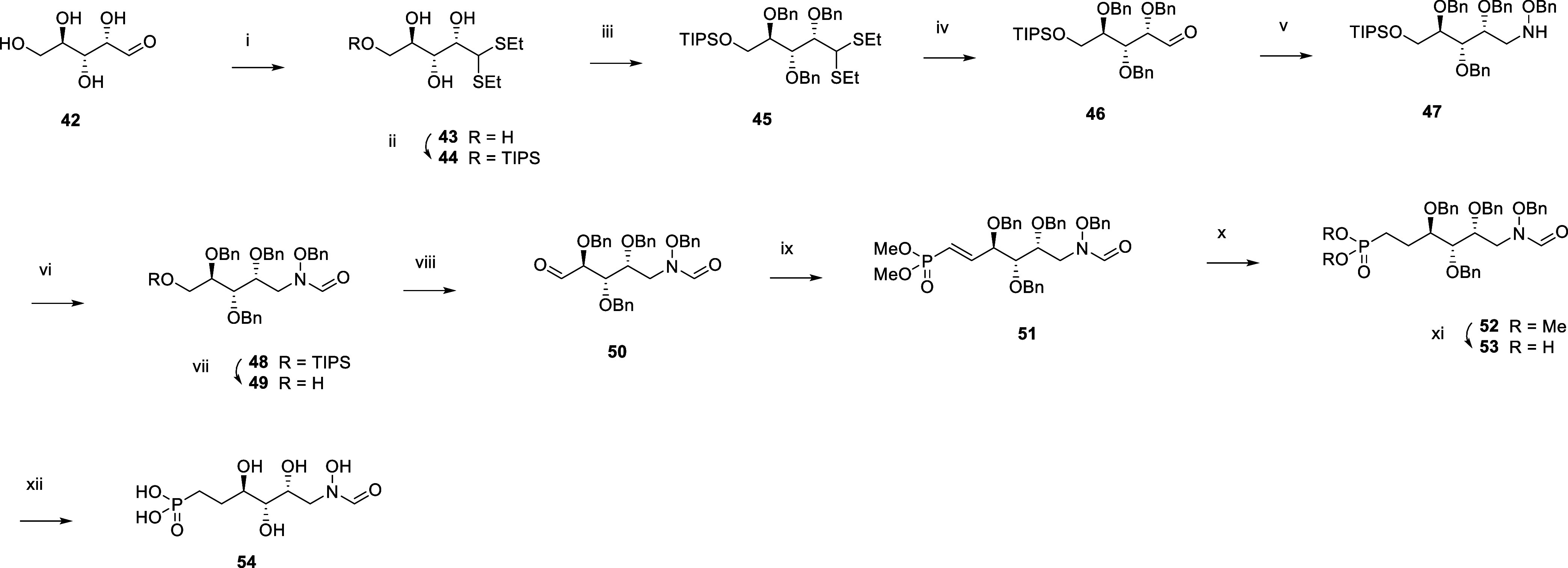
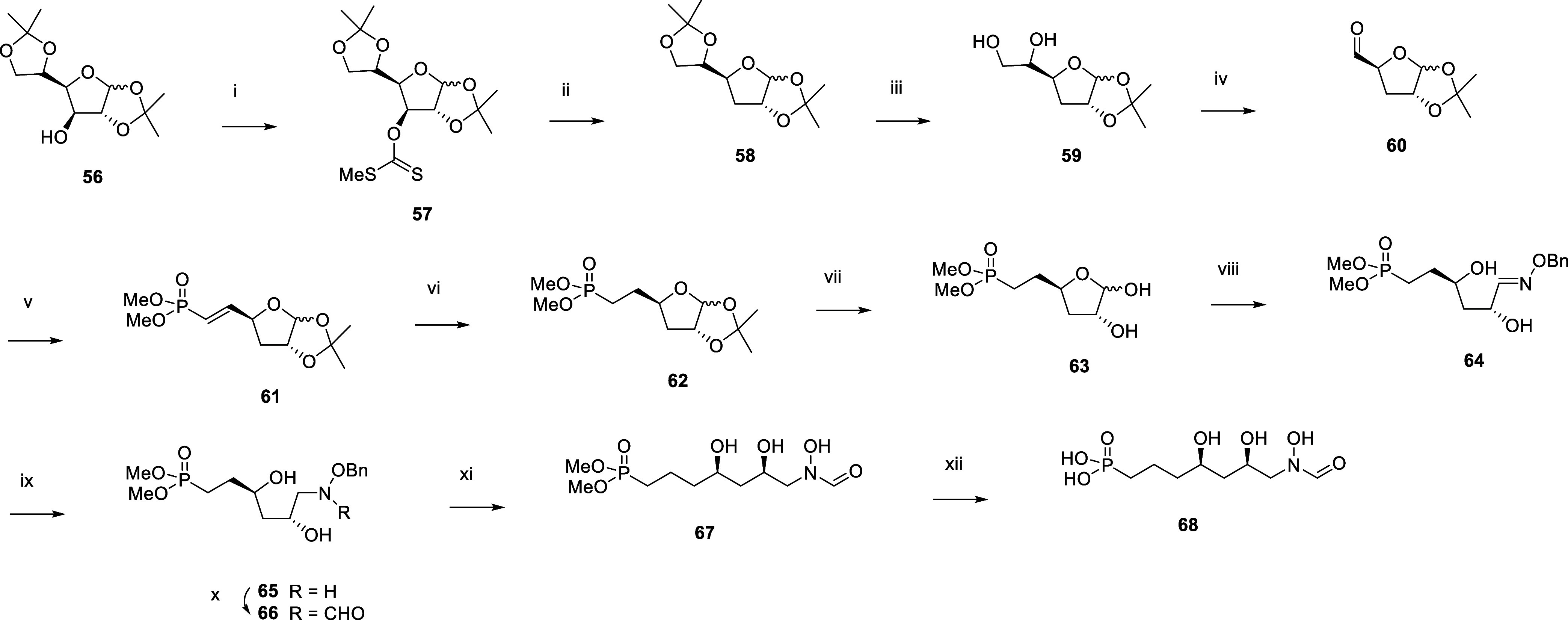
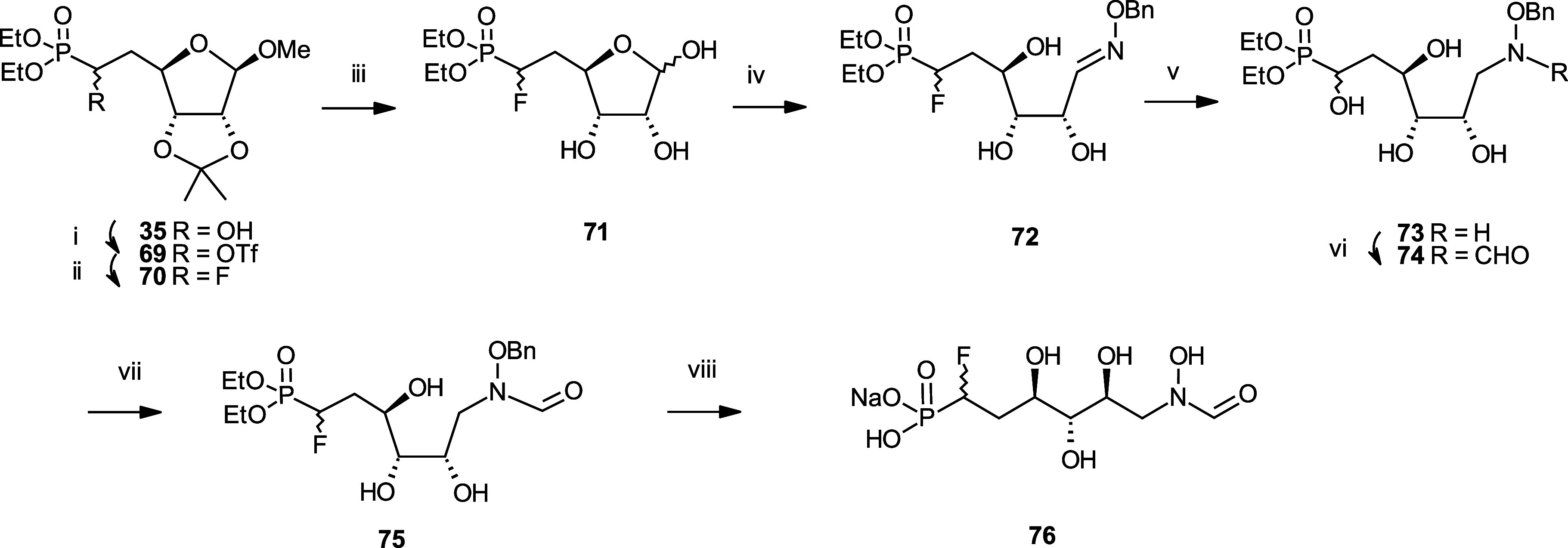
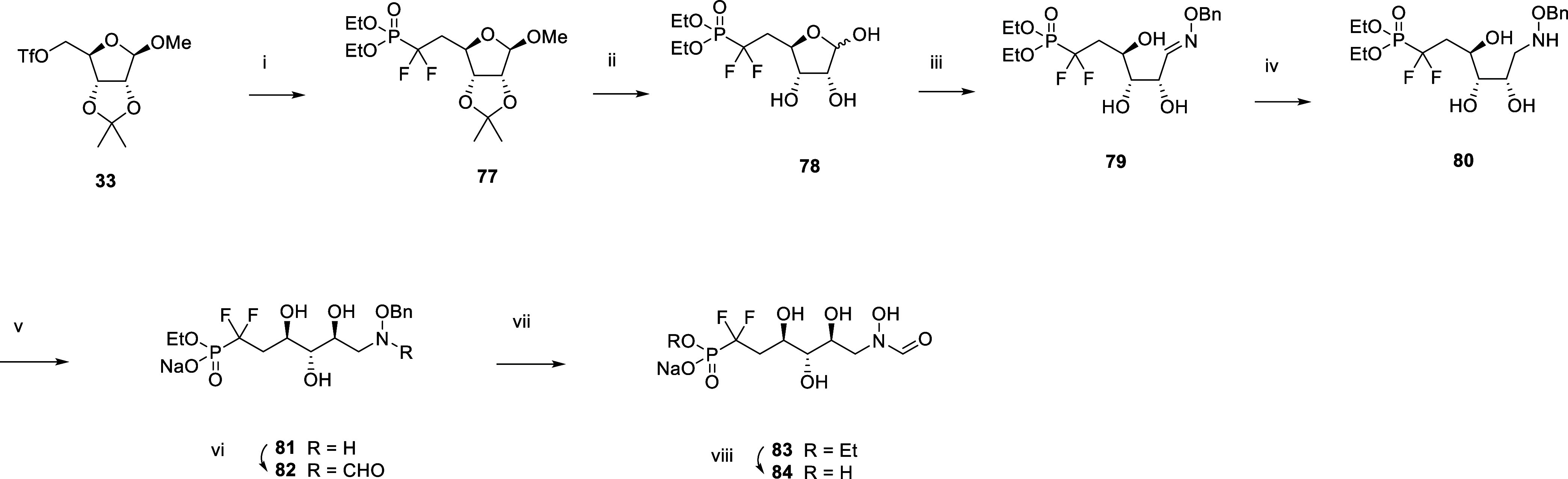
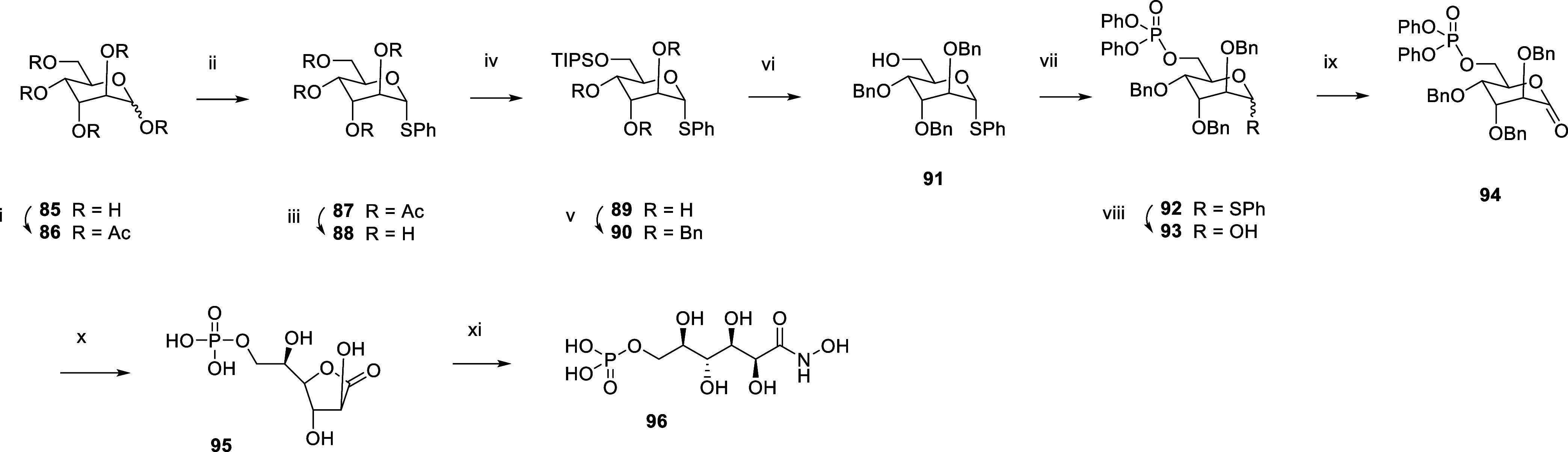
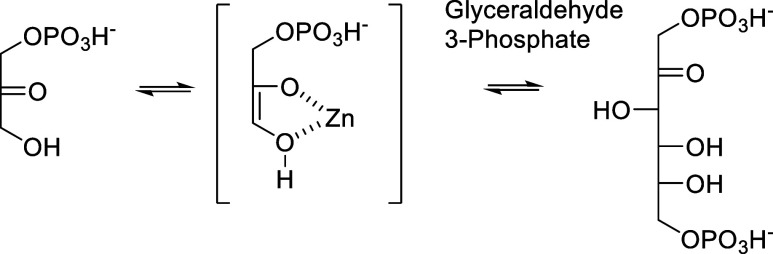
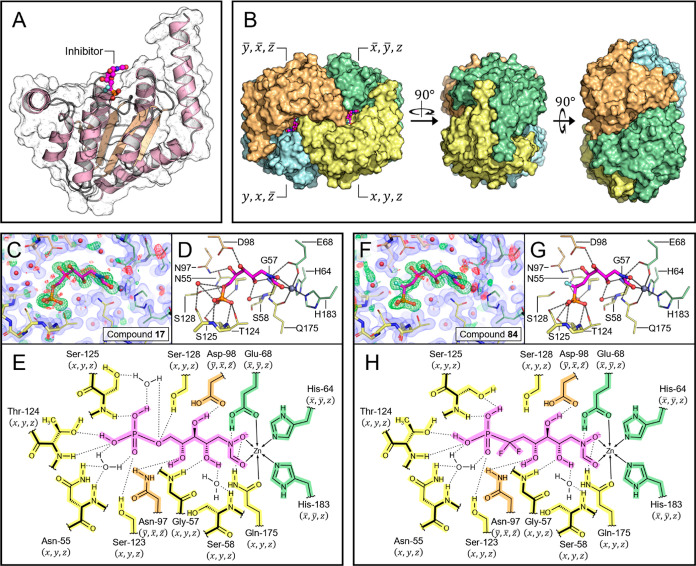
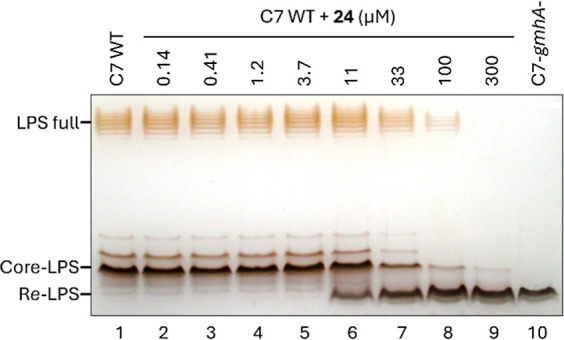
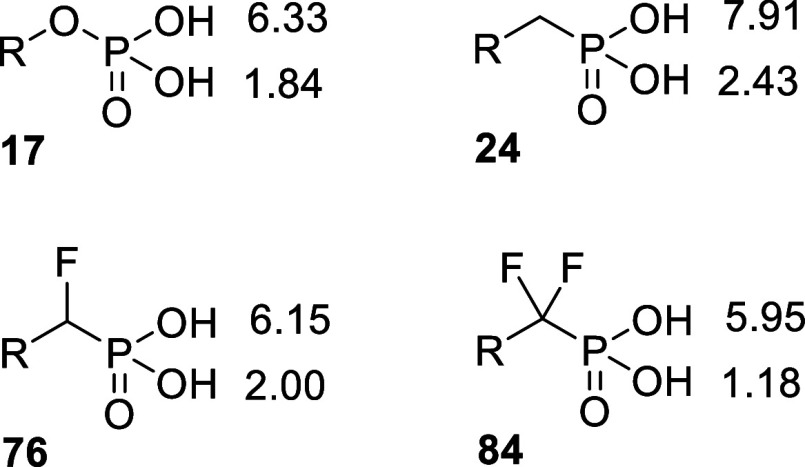
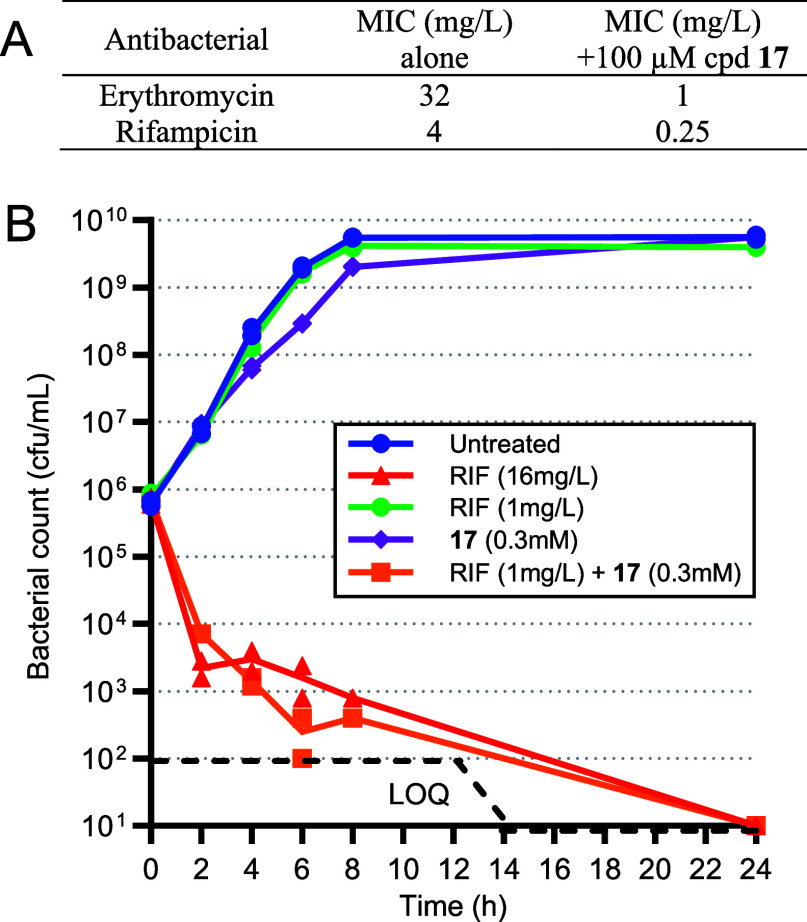
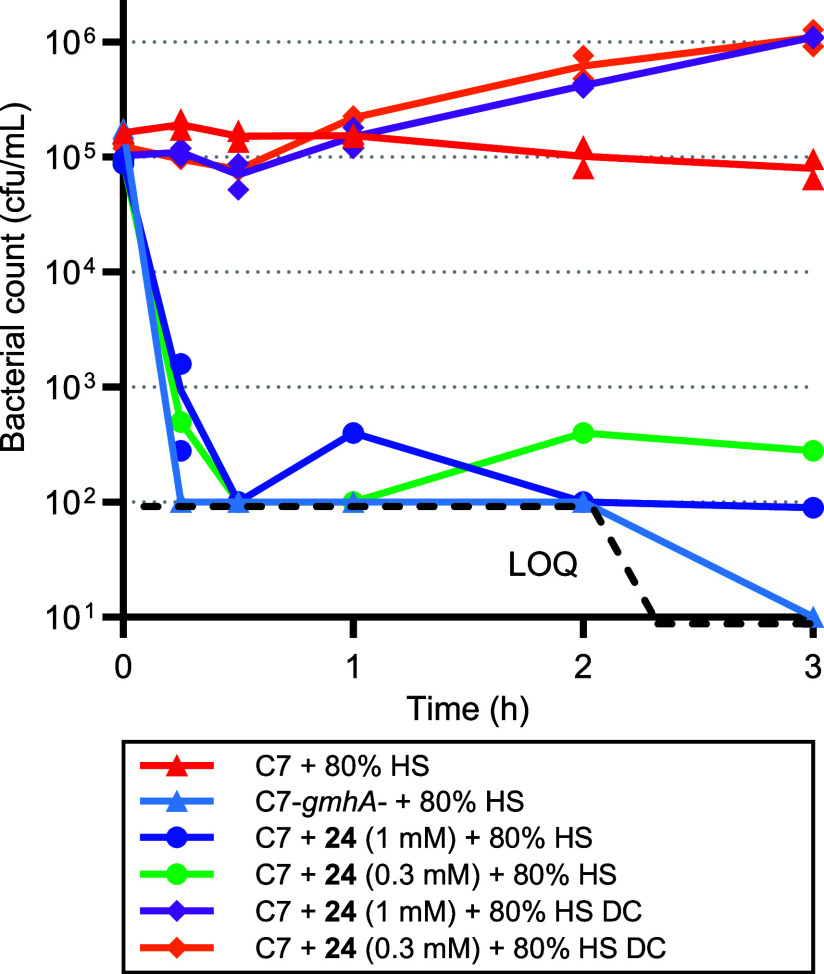
Inhibition of the biosynthesis of bacterial heptoses opens novel perspectives for antimicrobial therapies. The enzyme GmhA responsible for the first committed biosynthetic step catalyzes the conversion of sedoheptulose 7-phosphate into d-glycero-d-manno-heptose 7-phosphate and harbors a Zn2+ ion in the active site. A series of phosphoryl- and phosphonyl-substituted derivatives featuring a hydroxamate moiety were designed and prepared from suitably protected ribose or hexose derivatives. High-resolution crystal structures of GmhA complexed to two N-formyl hydroxamate inhibitors confirmed the binding interactions to a central Zn2+ ion coordination site. Some of these compounds were found to be nanomolar inhibitors of GmhA. While devoid of HepG2 cytotoxicity and antibacterial activity of their own, they demonstrated in vitro lipopolysaccharide heptosylation inhibition in Enterobacteriaceae as well as the potentiation of erythromycin and rifampicin in a wild-type Escherichia coli strain. These inhibitors pave the way for a novel treatment of Gram-negative infections.
Conflict of interest statement
The authors declare the following competing financial interest(s): When this study was performed F.M., D.A., S.F., D.B., and V.G. were Mutabilis full-time employees; M.Bl., M.Ba., M.H., and N.M.X. were full-time employees of the University of Natural Resources and Life Sciences, Vienna, Austria (funded by MUTABILIS, project no. 8389); J.M., E.A., and V.H. were Activation full-time employees, and F.L. was a full-time employee of Carbosynth.
Figures
References
-
- Fischbach M. A.; Walsh C. T. Antibiotics for emerging pathogens. Science 2009, 325, 1089–1093. 10.1126/science.1176667. - DOI - PMC - PubMed
- Chellat M. F.; Raguž L.; Riedl R. Targeting antibiotic resistance. Angew. Chem., Int. Ed. 2016, 55, 6600–6626. 10.1002/anie.201506818. - DOI - PMC - PubMed
- Culyba M. J.; Mo C. Y.; Kohli R. M. Targets for combating the evolution of acquired antibiotic resistance. Biochemistry 2015, 54, 3573–3582. 10.1021/acs.biochem.5b00109. - DOI - PMC - PubMed
- Brown E. D.; Wright G. D. Antibacterial drug discovery in the resistance era. Nature 2016, 529, 336–343. 10.1038/nature17042. - DOI - PubMed
- Taylor P. L.; Wright G. D. Novel approaches to discovery of antibacterial agents. Anim. Health Res. Rev. 2008, 9, 237–246. 10.1017/S1466252308001527. - DOI - PubMed
- Beyer P.; Paulin S. The antibacterial research and development pipeline needs urgent solutions. ACS Infect. Dis. 2020, 6, 1289–1291. 10.1021/acsinfecdis.0c00044. - DOI
-
- Raetz C. R.; Whitfield C. Lipopolysaccharide endotoxins. Annu. Rev. Biochem. 2002, 71, 635–700. 10.1146/annurev.biochem.71.110601.135414. - DOI - PMC - PubMed
- Holst O.Structure of the lipopolysaccharide core region. In Bacterial lipopolysaccharides; Knirel Y. A., Valvano M., Eds.; Springer: Wien, NY, 2011, pp 21–39.
-
- Brooke J. S.; Valvano M. A. Molecular cloning of the Haemophilus influenzae gmhA (lpcA) gene encoding a phosphoheptose isomerase required for lipopolysaccharide biosynthesis. J. Bacteriol. 1996, 178, 3339–3341. 10.1128/jb.178.11.3339-3341.1996. - DOI - PMC - PubMed
- Loutet S. A.; Flannagan R. S.; Kooi C.; Sokol P. A.; Valvano M. A. A complete lipopolysaccharide inner core oligosaccharide is required for resistance of Burkholderia cenocepacia to antimicrobial peptides and bacterial survival in vivo. J. Bacteriol. 2006, 188, 2073–2080. 10.1128/jb.188.6.2073-2080.2006. - DOI - PMC - PubMed
- Vaara M. Antibiotic-supersusceptible mutants of Escherichia coli and Salmonella typhimurium. Antimicrob. Agents Chemother. 1993, 37, 2255–2260. 10.1128/AAC.37.11.2255. - DOI - PMC - PubMed
-
- Escaich S. Antivirulence as a new antibacterial approach for chemotherapy. Curr. Opin. Chem. Biol. 2008, 12, 400–408. 10.1016/j.cbpa.2008.06.022. - DOI - PubMed
- Kuo C. J.; Chen J. W.; Chiu H. C.; Teng C. H.; Hsu T. I.; Lu P. J.; Syu W. Jr.; Wang S. T.; Chou T. C.; Chen C. S. Mutation of the enterohemorrhagic Escherichia coli core LPS biosynthesis enzyme rfaD confers hypersusceptibility to host intestinal innate immunity in vivo. Frontiers Cell. Infect. Microbiol. 2016, 6, 82.10.3389/fcimb.2016.00082. - DOI - PMC - PubMed
- Clatworthy A. E.; Pierson E.; Hung D. T. Targeting virulence: a new paradigm for antimicrobial therapy. Nat. Chem. Biol. 2007, 3, 541–548. 10.1038/nchembio.2007.24. - DOI - PubMed
-
- Eidels L.; Osborn M. J. Lipopolysaccharide and aldoheptose biosynthesis in transketolase mutants of Salmonella typhimurium. Proc. Natl. Acad. Sci. U.S.A. 1971, 68, 1673–1677. 10.1073/pnas.68.8.1673. - DOI - PMC - PubMed
- Chiu S.-F.; Teng K.-W.; Wang P.-C.; Chung H.-Y.; Wang C.-J.; Cheng H.-C.; Kao M.-C. Helicobacter pylori GmhB enzyme involved in ADP-heptose biosynthesis pathway is essential for lipopolysaccharide biosynthesis and bacterial virulence. Virulence 2021, 12, 1610–1628. 10.1080/21505594.2021.1938449. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
