

The genomic evolutionary dynamics and global circulation patterns of respiratory syncytial virus
- PMID: 38600104
- PMCID: PMC11006891
- DOI: 10.1038/s41467-024-47118-6
The genomic evolutionary dynamics and global circulation patterns of respiratory syncytial virus
Abstract
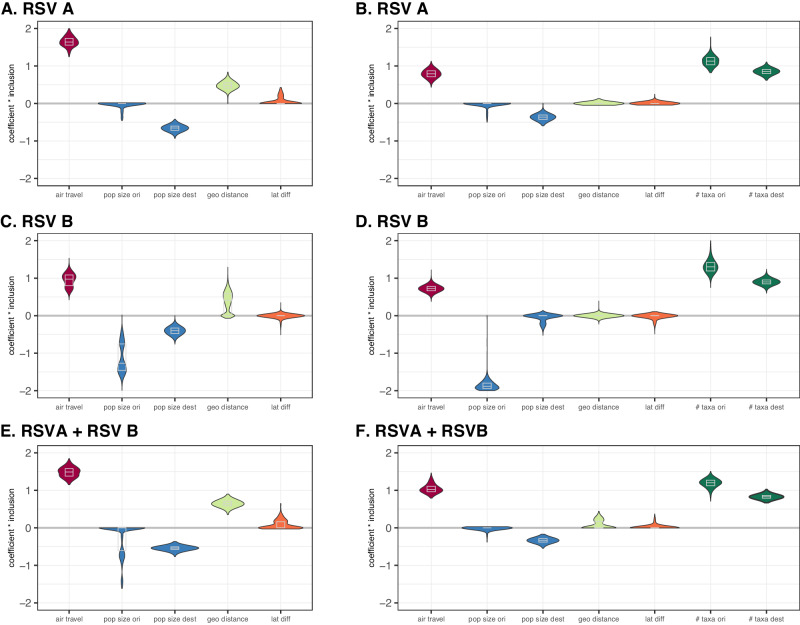
Respiratory syncytial virus (RSV) is a leading cause of acute lower respiratory tract infection in young children and the second leading cause of infant death worldwide. While global circulation has been extensively studied for respiratory viruses such as seasonal influenza, and more recently also in great detail for SARS-CoV-2, a lack of global multi-annual sampling of complete RSV genomes limits our understanding of RSV molecular epidemiology. Here, we capitalise on the genomic surveillance by the INFORM-RSV study and apply phylodynamic approaches to uncover how selection and neutral epidemiological processes shape RSV diversity. Using complete viral genome sequences, we show similar patterns of site-specific diversifying selection among RSVA and RSVB and recover the imprint of non-neutral epidemic processes on their genealogies. Using a phylogeographic approach, we provide evidence for air travel governing the global patterns of RSVA and RSVB spread, which results in a considerable degree of phylogenetic mixing across countries. Our findings highlight the potential of systematic global RSV genomic surveillance for transforming our understanding of global RSV spread.
© 2024. The Author(s).
Conflict of interest statement
L.J.B. has regular interaction with pharmaceutical and other industrial partners. He has not received personal fees or other personal benefits. U.M.C.U. has received major funding (>€100,000 per industrial partner) for investigator initiated studies from AbbVie, MedImmune, Janssen, the Bill and Melinda Gates Foundation, Nutricia (Danone) and MeMed Diagnostics. U.M.C.U. has received major cash or in kind funding as part of the public private partnership IMI-funded RESCEU project from GSK, Novavax, Janssen, AstraZeneca, Pfizer and Sanofi. U.M.C.U. has received major funding from Julius Clinical for participating in the INFORM-RSV study sponsored by AstraZeneca and Sanofi. U.M.C.U. has received minor funding for participation in trials by Regeneron and Janssen from 2015–2017 (total annual estimate less than €20,000). U.M.C.U. received minor funding for consultation and invited lectures by AbbVie, MedImmune, Ablynx, Bavaria Nordic, MabXience, Novavax, Pfizer, and Janssen (total annual estimate less than €20,000). L.J.B. is the founding chairman of the ReSViNET Foundation. P.L. and M.A.S. acknowledge support from the European Union’s Horizon 2020 research and innovation programme (grant agreement no. 725422-ReservoirDOCS), from the Wellcome Trust through project 206298/Z/17/Z and from the NIH grant R01 AI153044. P.L. acknowledges support from the Research Foundation - Flanders (‘Fonds voor Wetenschappelijk Onderzoek - Vlaanderen’, G0D5117N and G051322N) and from the European Union’s Horizon 2020 project MOOD (grant agreement no. 874850). D.W. and E.J.K. are employees of AstraZeneca. The remaining authors declare no competing interests.
Figures


References
-
- Langedijk, A. C. & Bont, L. J. Respiratory syncytial virus infection and novel interventions. Nat. Rev. Microbiol., 10.1038/s41579-023-00919-w (2023). - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
