

Synthetic biology approaches for improving the specificity and efficacy of cancer immunotherapy
- PMID: 38605087
- PMCID: PMC11061174
- DOI: 10.1038/s41423-024-01153-x
Synthetic biology approaches for improving the specificity and efficacy of cancer immunotherapy
Abstract
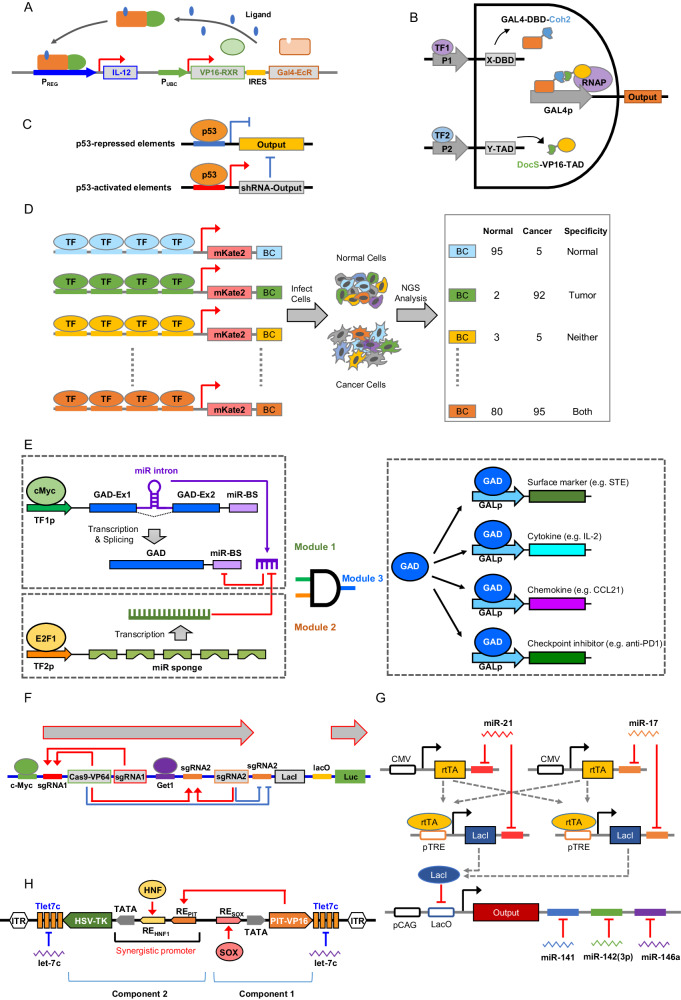
Immunotherapy has shown robust efficacy in treating a broad spectrum of hematological and solid cancers. Despite the transformative impact of immunotherapy on cancer treatment, several outstanding challenges remain. These challenges include on-target off-tumor toxicity, systemic toxicity, and the complexity of achieving potent and sustainable therapeutic efficacy. Synthetic biology has emerged as a promising approach to overcome these obstacles, offering innovative tools for engineering living cells with customized functions. This review provides an overview of the current landscape and future prospects of cancer immunotherapy, particularly emphasizing the role of synthetic biology in augmenting its specificity, controllability, and efficacy. We delineate and discuss two principal synthetic biology strategies: those targeting tumor surface antigens with engineered immune cells and those detecting intratumoral disease signatures with engineered gene circuits. This review concludes with a forward-looking perspective on the enduring challenges in cancer immunotherapy and the potential breakthroughs that synthetic biology may contribute to the field.
Keywords: Adoptive cell therapy.; Cancer immunotherapy; Gene circuit; Gene therapy; Synthetic biology.
© 2024. The Author(s).
Conflict of interest statement
MW is the inventor and holds several patents related to a cancer-targeting gene circuit platform described in the review. Other authors declare no competing interests.
Figures
Similar articles
-
Engineering advanced cancer therapies with synthetic biology.Nat Rev Cancer. 2019 Apr;19(4):187-195. doi: 10.1038/s41568-019-0121-0. Nat Rev Cancer. 2019. PMID: 30837696 Review.
-
A synthetic biology approach to engineering circuits in immune cells.Immunol Rev. 2023 Nov;320(1):120-137. doi: 10.1111/imr.13244. Epub 2023 Jul 19. Immunol Rev. 2023. PMID: 37464881 Review.
-
Engineering Microorganisms for Cancer Immunotherapy.Adv Healthc Mater. 2024 Jul;13(18):e2304649. doi: 10.1002/adhm.202304649. Epub 2024 Apr 21. Adv Healthc Mater. 2024. PMID: 38598792 Review.
-
Synthetic biology approaches for enhancing safety and specificity of CAR-T cell therapies for solid cancers.Cytotherapy. 2024 Aug;26(8):842-857. doi: 10.1016/j.jcyt.2024.03.484. Epub 2024 Mar 30. Cytotherapy. 2024. PMID: 38639669 Review.
-
Synthetic biology approaches in cancer immunotherapy, genetic network engineering, and genome editing.Integr Biol (Camb). 2016 Apr 18;8(4):504-17. doi: 10.1039/c5ib00325c. Epub 2016 Apr 12. Integr Biol (Camb). 2016. PMID: 27068224 Review.
Cited by
-
Next-Generation Vaccine Platforms: Integrating Synthetic Biology, Nanotechnology, and Systems Immunology for Improved Immunogenicity.Vaccines (Basel). 2025 May 30;13(6):588. doi: 10.3390/vaccines13060588. Vaccines (Basel). 2025. PMID: 40573919 Free PMC article. Review.
-
Opportunities to Modulate Tumor Ecosystem Toward Successful Glioblastoma Immunotherapy.Cancer Sci. 2025 Jun;116(6):1482-1499. doi: 10.1111/cas.70052. Epub 2025 Mar 23. Cancer Sci. 2025. PMID: 40123277 Free PMC article. Review.
-
Cancer stem cells: landscape, challenges and emerging therapeutic innovations.Signal Transduct Target Ther. 2025 Aug 5;10(1):248. doi: 10.1038/s41392-025-02360-2. Signal Transduct Target Ther. 2025. PMID: 40759634 Free PMC article. Review.
-
"Lost in translation?" Animal research in the era of precision medicine.J Transl Med. 2025 Feb 4;23(1):152. doi: 10.1186/s12967-025-06084-3. J Transl Med. 2025. PMID: 39905446 Free PMC article. Review.
-
Cell-based immunotherapies for solid tumors: advances, challenges, and future directions.Front Oncol. 2025 Apr 28;15:1551583. doi: 10.3389/fonc.2025.1551583. eCollection 2025. Front Oncol. 2025. PMID: 40356763 Free PMC article. Review.
References
-
- Couzin-Frankel J. Breakthrough of the year 2013. Cancer immunotherapy. Science. 2013;342:1432–3. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
