

Single-molecule epitranscriptomic analysis of full-length HIV-1 RNAs reveals functional roles of site-specific m6As
- PMID: 38605174
- PMCID: PMC11087264
- DOI: 10.1038/s41564-024-01638-5
Single-molecule epitranscriptomic analysis of full-length HIV-1 RNAs reveals functional roles of site-specific m6As
Abstract
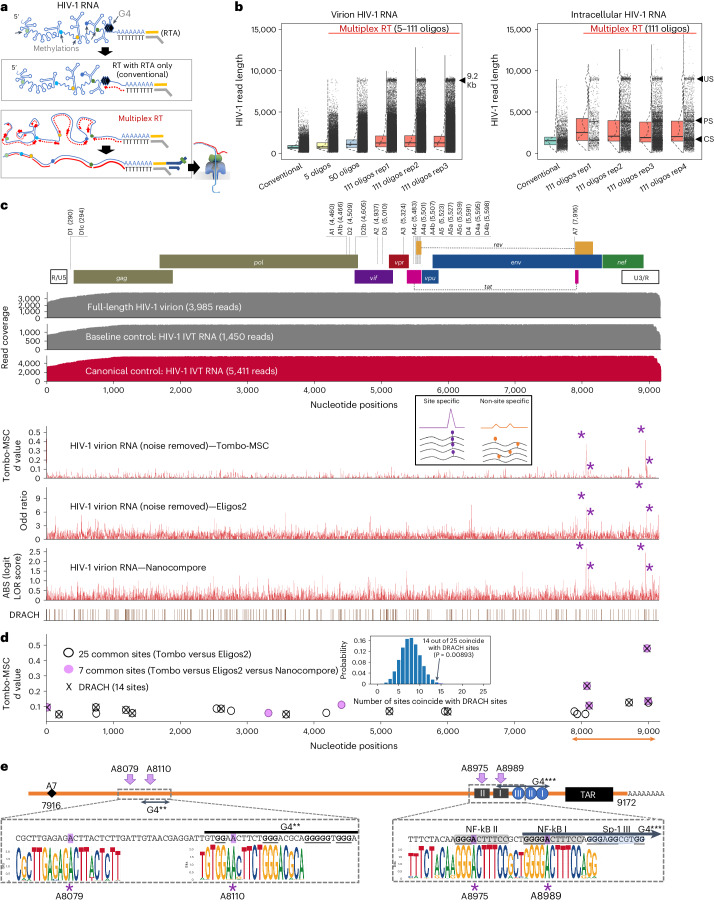
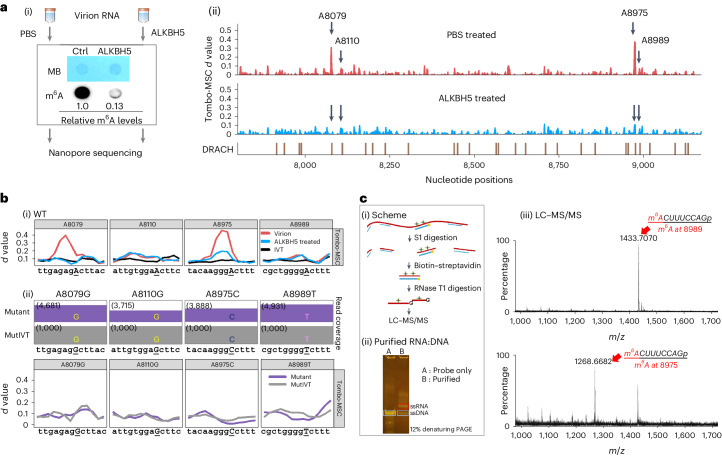
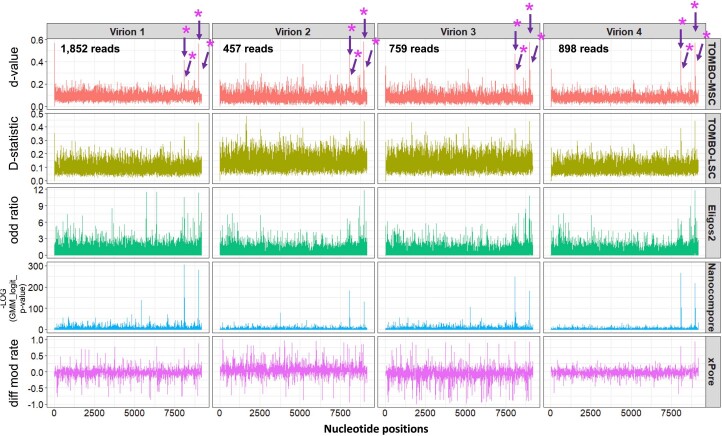
Although the significance of chemical modifications on RNA is acknowledged, the evolutionary benefits and specific roles in human immunodeficiency virus (HIV-1) replication remain elusive. Most studies have provided only population-averaged values of modifications for fragmented RNAs at low resolution and have relied on indirect analyses of phenotypic effects by perturbing host effectors. Here we analysed chemical modifications on HIV-1 RNAs at the full-length, single RNA level and nucleotide resolution using direct RNA sequencing methods. Our data reveal an unexpectedly simple HIV-1 modification landscape, highlighting three predominant N6-methyladenosine (m6A) modifications near the 3' end. More densely installed in spliced viral messenger RNAs than in genomic RNAs, these m6As play a crucial role in maintaining normal levels of HIV-1 RNA splicing and translation. HIV-1 generates diverse RNA subspecies with distinct m6A ensembles, and maintaining multiple of these m6As on its RNAs provides additional stability and resilience to HIV-1 replication, suggesting an unexplored viral RNA-level evolutionary strategy.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures













References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
