

Expanding SPG18 clinical spectrum: autosomal dominant mutation causes complicated hereditary spastic paraplegia in a large family
- PMID: 38607533
- PMCID: PMC11306645
- DOI: 10.1007/s10072-024-07500-0
Expanding SPG18 clinical spectrum: autosomal dominant mutation causes complicated hereditary spastic paraplegia in a large family
Abstract
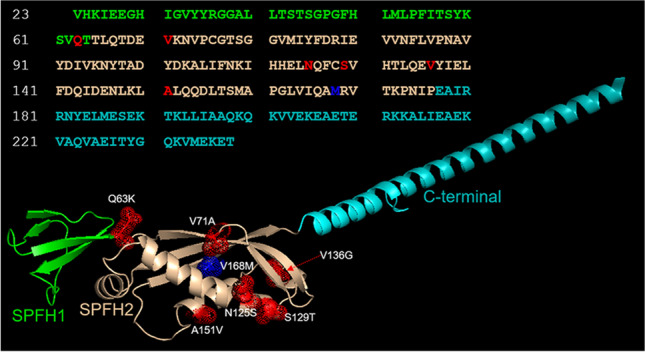
Background: SPG18 is caused by mutations in the endoplasmic reticulum lipid raft associated 2 (ERLIN2) gene. Autosomal recessive (AR) mutations are usually associated with complicated hereditary spastic paraplegia (HSP), while autosomal dominant (AD) mutations use to cause pure SPG18.
Aim: To define the variegate clinical spectrum of the SPG18 and to evaluate a dominant negative effect of erlin2 (encoded by ERLIN2) on oligomerization as causing differences between AR and AD phenotypes.
Methods: In a four-generation pedigree with an AD pattern, a spastic paraplegia multigene panel test was performed. Oligomerization of erlin2 was analyzed with velocity gradient assay in fibroblasts of the proband and healthy subjects.
Results: Despite the common p.V168M mutation identified in ERLIN2, a phenoconversion to amyotrophic lateral sclerosis (ALS) was observed in the second generation, pure HSP in the third generation, and a complicated form with psychomotor delay and epilepsy in the fourth generation. Erlin2 oligomerization was found to be normal.
Discussion: We report the first AD SPG18 family with a complicated phenotype, and we ruled out a dominant negative effect of V168M on erlin2 oligomerization. Therefore, our data do not support the hypothesis of a relationship between the mode of inheritance and the phenotype, but confirm the multifaceted nature of SPG18 on both genetic and clinical point of view. Clinicians should be aware of the importance of conducting an in-depth clinical evaluation to unmask all the possible manifestations associated to an only apparently pure SPG18 phenotype. We confirm the genotype-phenotype correlation between V168M and ALS emphasizing the value of close follow-up.
Keywords: Amyotrophic lateral sclerosis (ALS); ERLIN2; Hereditary spastic paraplegia (HSP); SPG18.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures


References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
