

Spatially resolved immune exhaustion within the alloreactive microenvironment predicts liver transplant rejection
- PMID: 38608023
- PMCID: PMC11014454
- DOI: 10.1126/sciadv.adm8841
Spatially resolved immune exhaustion within the alloreactive microenvironment predicts liver transplant rejection
Abstract
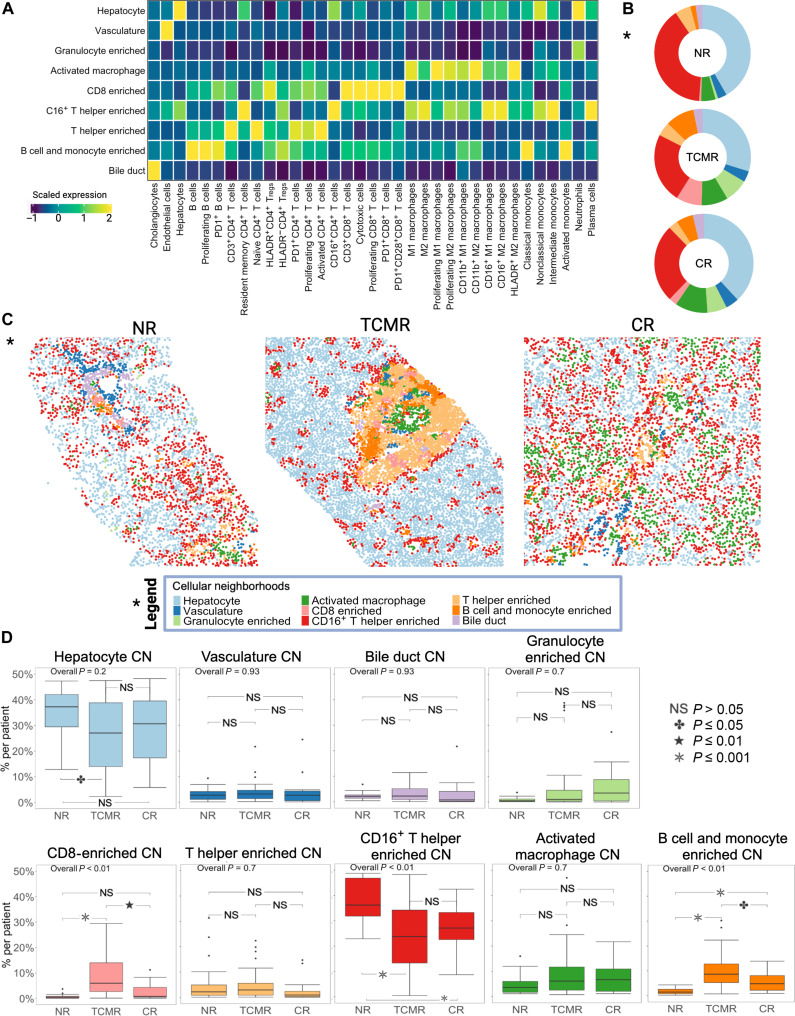
Allograft rejection is common following clinical organ transplantation, but defining specific immune subsets mediating alloimmunity has been elusive. Calcineurin inhibitor dose escalation, corticosteroids, and/or lymphocyte depleting antibodies have remained the primary options for treatment of clinical rejection episodes. Here, we developed a highly multiplexed imaging mass cytometry panel to study the immune response in archival biopsies from 79 liver transplant (LT) recipients with either no rejection (NR), acute T cell-mediated rejection (TCMR), or chronic rejection (CR). This approach generated a spatially resolved proteomic atlas of 461,816 cells (42 phenotypes) derived from 96 pathologist-selected regions of interest. Our analysis revealed that regulatory (HLADR+ Treg) and PD1+ T cell phenotypes (CD4+ and CD8+ subsets), combined with variations in M2 macrophage polarization, were a unique signature of active TCMR. These data provide insights into the alloimmune microenvironment in clinical LT, including identification of potential targets for focused immunotherapy during rejection episodes and suggestion of a substantial role for immune exhaustion in TCMR.
Figures







Update of
-
Spatially resolved immune exhaustion within the alloreactive microenvironment predicts liver transplant rejection.Res Sq [Preprint]. 2023 Jul 3:rs.3.rs-3044385. doi: 10.21203/rs.3.rs-3044385/v1. Res Sq. 2023. Update in: Sci Adv. 2024 Apr 12;10(15):eadm8841. doi: 10.1126/sciadv.adm8841. PMID: 37461437 Free PMC article. Updated. Preprint.
References
-
- Shaked A., Ghobrial R. M., Merion R. M., Shearon T. H., Emond J. C., Fair J. H., Fisher R. A., Kulik L. M., Pruett T. L., Terrault N. A., A2ALL Study Group , Incidence and severity of acute cellular rejection in recipients undergoing adult living donor or deceased donor liver transplantation. Am. J. Transplant. 9, 301–308 (2009). - PMC - PubMed
-
- Bartlett A. S., Ramadas R., Furness S., Gane E., McCall J. L., The natural history of acute histologic rejection without biochemical graft dysfunction in orthotopic liver transplantation: A systematic review. Liver Transpl. 8, 1147–1153 (2002). - PubMed
-
- Levitsky J., Goldberg D., Smith A. R., Mansfield S. A., Gillespie B. W., Merion R. M., Lok A. S. F., Levy G., Kulik L., Abecassis M., Shaked A., Acute rejection increases risk of graft failure and death in recent liver transplant recipients. Clin. Gastroenterol. Hepatol. 15, 584–593.e2 (2017). - PMC - PubMed
-
- Demetris A. J., Batts K. P., Dhillon A. P., Ferrell L., Fung J., Geller S. A., Hart J., Hayry P., Hofmann W. J., Hubscher S., Kemnitz J., Koukoulis G., Lee R. G., Lewin K. J., Ludwig J., Markin R. S., Petrovic L. M., Phillips M. J., Portmann B., Rakela J., Randhawa P., Reinholt F. P., Reyne’s M., Reyne’s R., Robert M., Schlitt H., Solez K., Snover D., Taskinen E., Thung S. N., Tillery G. W., Wiesner R. H., Wight D. G. D., Williams J. W., Yamabe H., Banff Schema for grading liver allograft rejection: An international consensus document. Hepatology 23, 658–663 (1997). - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
