

Splice modulators target PMS1 to reduce somatic expansion of the Huntington's disease-associated CAG repeat
- PMID: 38609352
- PMCID: PMC11015039
- DOI: 10.1038/s41467-024-47485-0
Splice modulators target PMS1 to reduce somatic expansion of the Huntington's disease-associated CAG repeat
Abstract
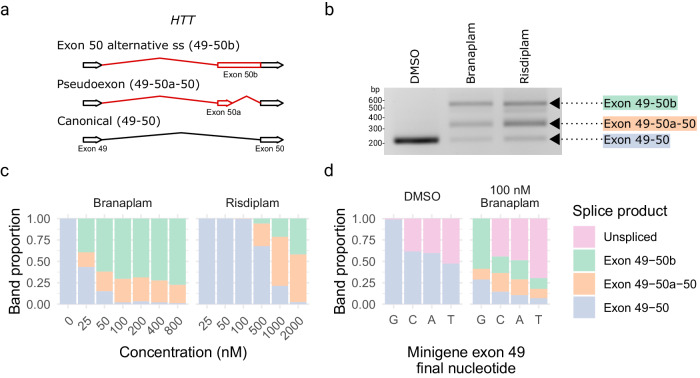
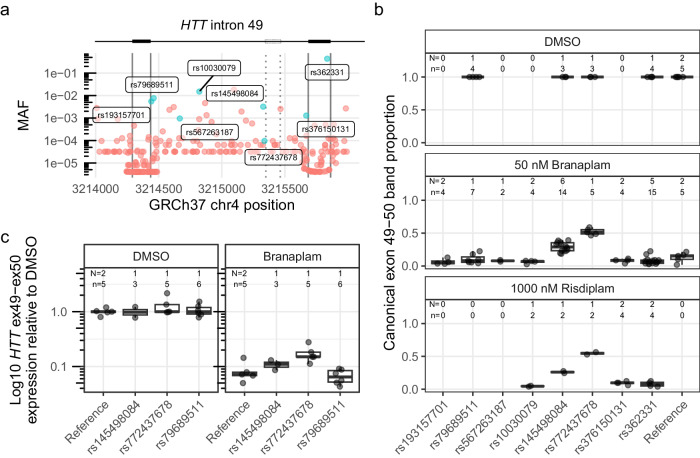
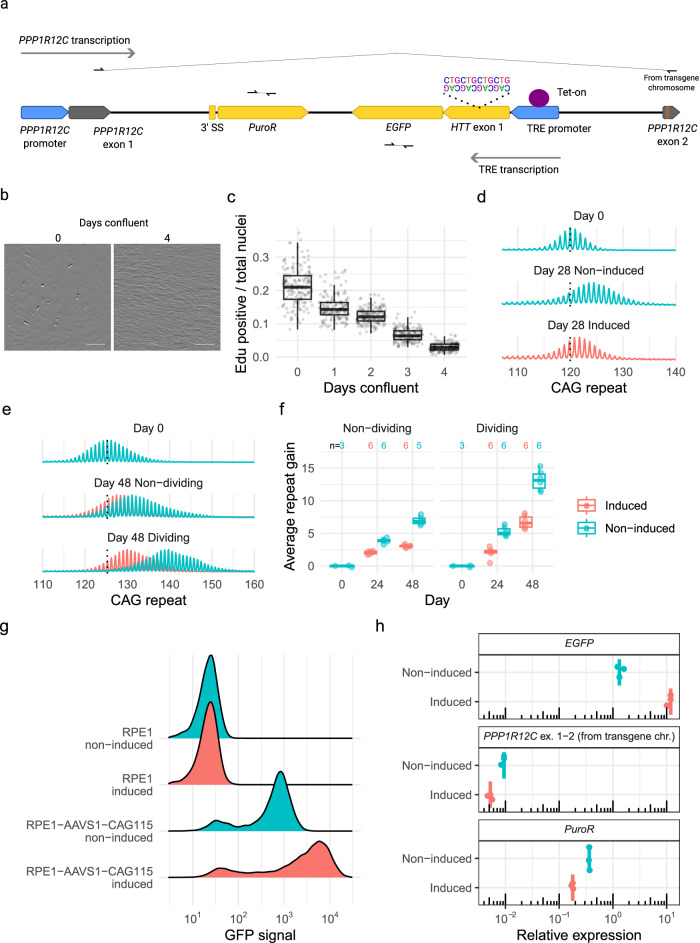
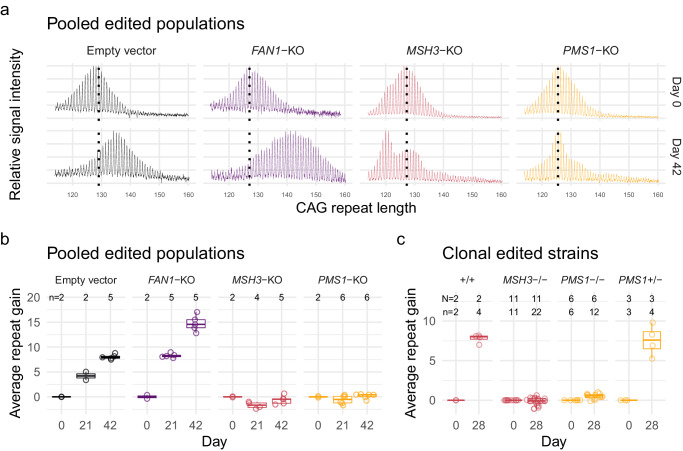
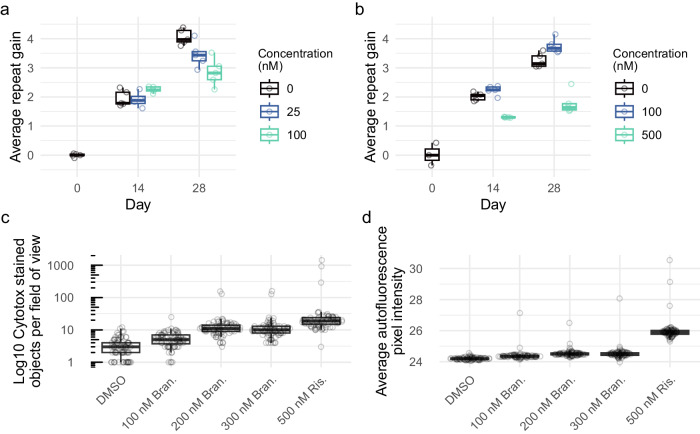
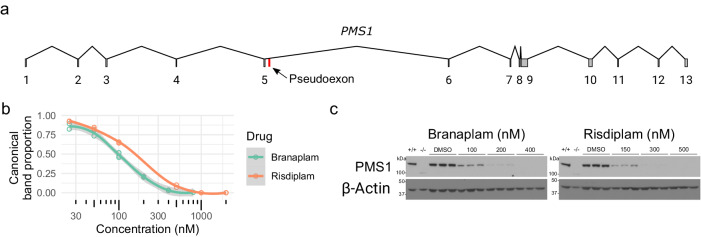
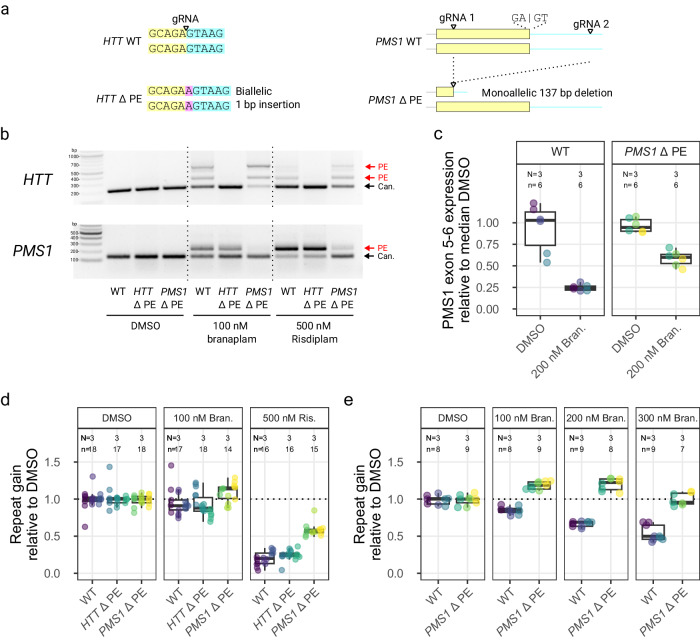
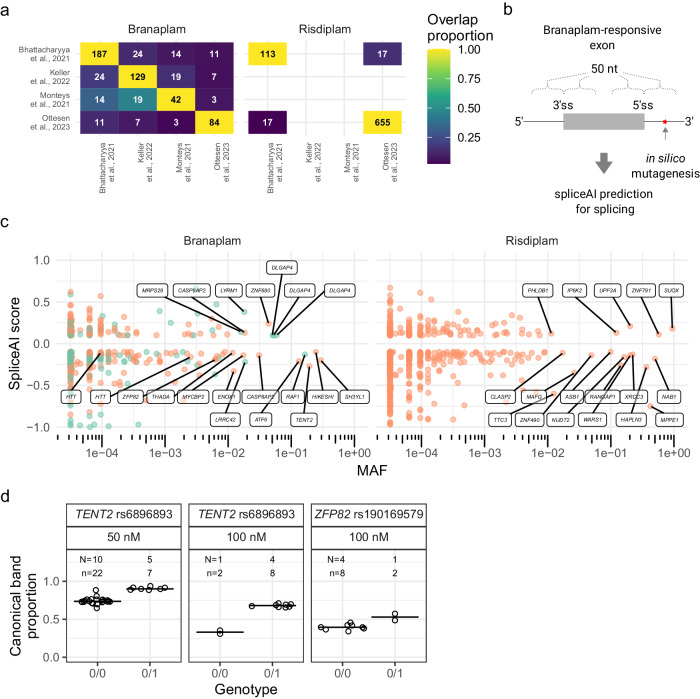
Huntington's disease (HD) is a dominant neurological disorder caused by an expanded HTT exon 1 CAG repeat that lengthens huntingtin's polyglutamine tract. Lowering mutant huntingtin has been proposed for treating HD, but genetic modifiers implicate somatic CAG repeat expansion as the driver of onset. We find that branaplam and risdiplam, small molecule splice modulators that lower huntingtin by promoting HTT pseudoexon inclusion, also decrease expansion of an unstable HTT exon 1 CAG repeat in an engineered cell model. Targeted CRISPR-Cas9 editing shows this effect is not due to huntingtin lowering, pointing instead to pseudoexon inclusion in PMS1. Homozygous but not heterozygous inactivation of PMS1 also reduces CAG repeat expansion, supporting PMS1 as a genetic modifier of HD and a potential target for therapeutic intervention. Although splice modulation provides one strategy, genome-wide transcriptomics also emphasize consideration of cell-type specific effects and polymorphic variation at both target and off-target sites.
© 2024. The Author(s).
Conflict of interest statement
J.F.G. and V.C.W. were founding scientific advisory board members with a financial interest in Triplet Therapeutics Inc. Their financial interests were reviewed and are managed by Massachusetts General Hospital (MGH) and Mass General Brigham (MGB) in accordance with their conflict of interest policies. J.F.G. consults for Transine Therapeutics, Inc. (dba Harness Therapeutics) and has previously provided paid consulting services to Wave Therapeutics USA Inc., Biogen Inc. and Pfizer Inc. V.C.W. is a scientific advisory board member of LoQus23 Therapeutics Ltd. and has provided paid consulting services to Acadia Pharmaceuticals Inc., Alnylam Inc., Biogen Inc., Passage Bio and Rgenta Therapeutics. R.M.P. and V.C.W. have received research support from Pfizer Inc. B.P.K. is a consultant for EcoR1 capital and Novartis Venture Fund, and is on the scientific advisory board of Acrigen Biosciences, Life Edit Therapeutics and Prime Medicine. B.P.K. has a financial interest in Prime Medicine, Inc., a company developing therapeutic CRISPR-Cas technologies for gene editing. B.P.K.‘s interests were reviewed and are managed by MGH and MGB in accordance with their conflict-of-interest policies. J-M.L. consults for Life Edit Therapeutics and serves on the scientific advisory board of GenEdit Inc. E.M. is inventor on an International Patent Application Number PCT/US2021/012103, assigned to Massachusetts General Hospital and PTC Therapeutics entitled “RNA Splicing Modulation” related to use of BPN-15477 in modulating splicing. E.M. is a scientific advisory board member of ReviR Therapeutics, Inc and has received research support from PTC Therapeutics, Inc. The other authors declare no competing interests.
Figures
Update of
-
PMS1 as a target for splice modulation to prevent somatic CAG repeat expansion in Huntington's disease.bioRxiv [Preprint]. 2023 Jul 27:2023.07.25.550489. doi: 10.1101/2023.07.25.550489. bioRxiv. 2023. Update in: Nat Commun. 2024 Apr 12;15(1):3182. doi: 10.1038/s41467-024-47485-0. PMID: 37547003 Free PMC article. Updated. Preprint.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
