

Genetic Diversity of Avian Influenza Viruses Detected in Waterbirds in Northeast Italy Using Two Different Sampling Strategies
- PMID: 38612257
- PMCID: PMC11010841
- DOI: 10.3390/ani14071018
Genetic Diversity of Avian Influenza Viruses Detected in Waterbirds in Northeast Italy Using Two Different Sampling Strategies
Abstract
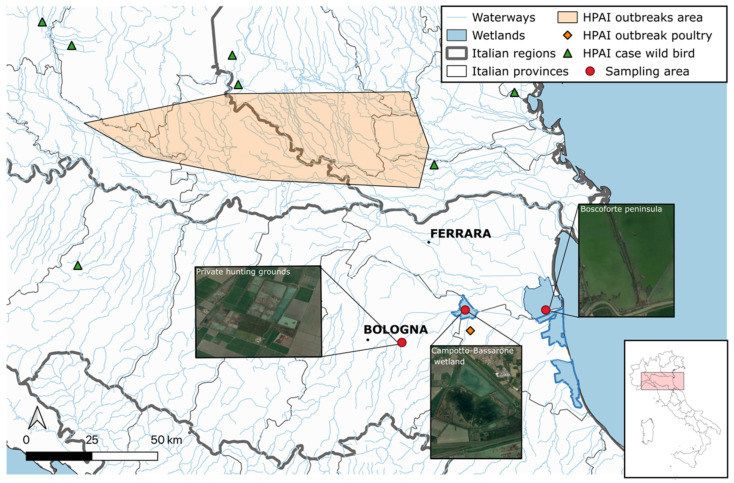
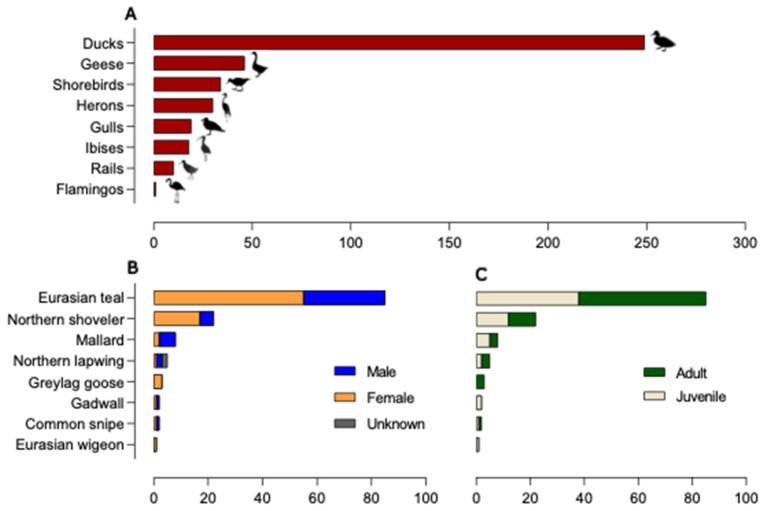
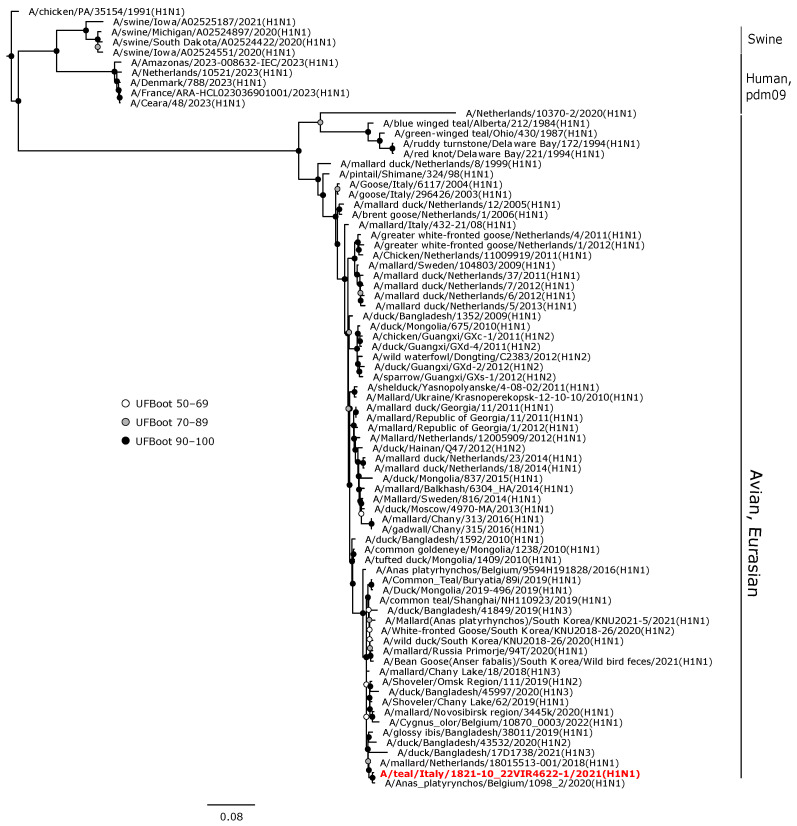
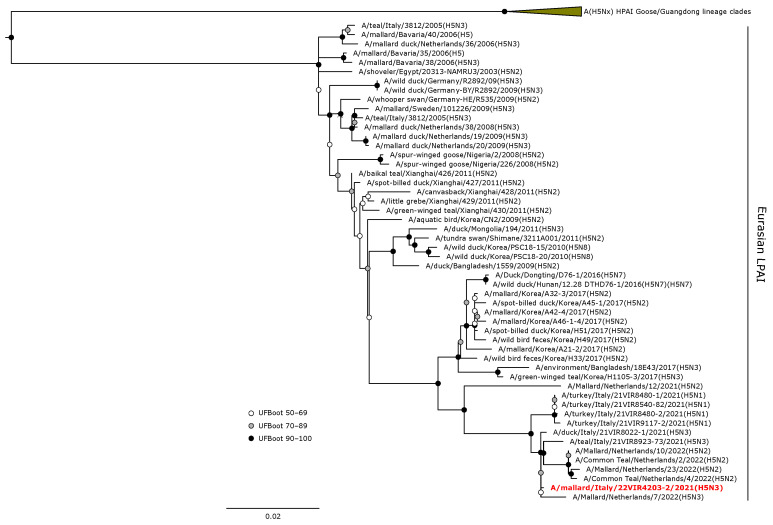
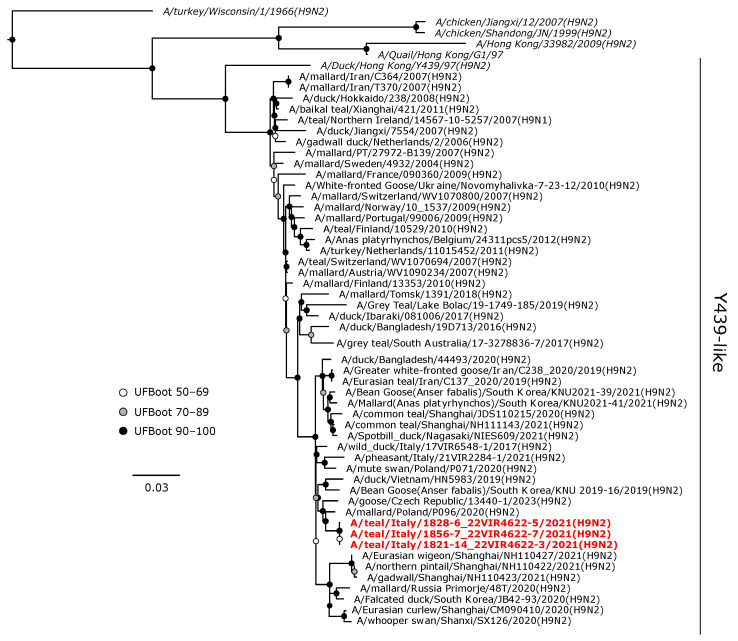
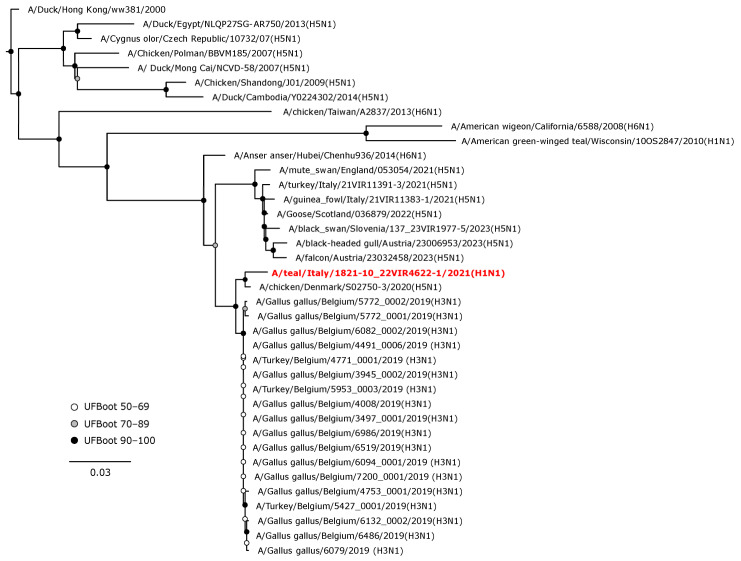
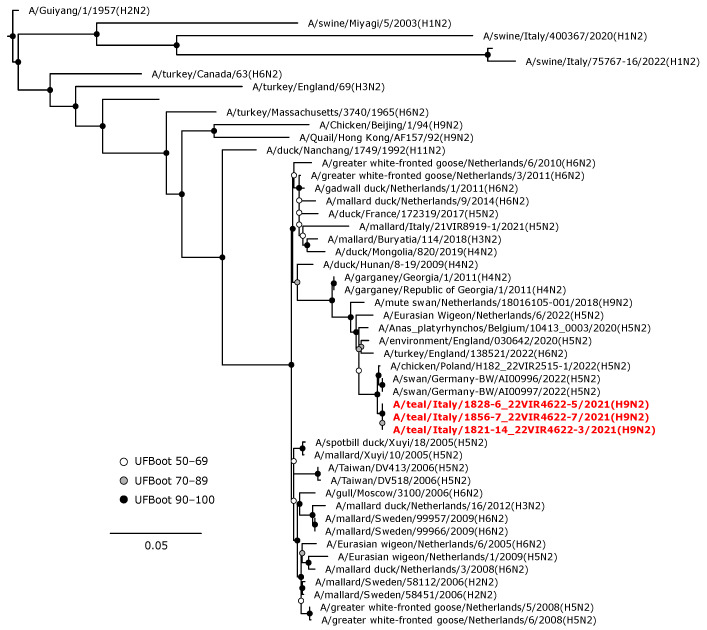
Avian influenza viruses (AIVs), which circulate endemically in wild aquatic birds, pose a significant threat to poultry and raise concerns for their zoonotic potential. From August 2021 to April 2022, a multi-site cross-sectional study involving active AIV epidemiological monitoring was conducted in wetlands of the Emilia-Romagna region, northern Italy, adjacent to densely populated poultry areas. A total of 129 cloacal swab samples (CSs) and 407 avian faecal droppings samples (FDs) were collected, with 7 CSs (5.4%) and 4 FDs (1%) testing positive for the AIV matrix gene through rRT-PCR. A COI-barcoding protocol was applied to recognize the species of origin of AIV-positive FDs. Multiple low-pathogenic AIV subtypes were identified, and five of these were isolated, including an H5N3, an H1N1, and three H9N2 in wild ducks. Following whole-genome sequencing, phylogenetic analyses of the hereby obtained strains showed close genetic relationships with AIVs detected in countries along the Black Sea/Mediterranean migratory flyway. Notably, none of the analyzed gene segments were genetically related to HPAI H5N1 viruses of clade 2.3.4.4b isolated from Italian poultry during the concurrent 2021-2022 epidemic. Overall, the detected AIV genetic diversity emphasizes the necessity for ongoing monitoring in wild hosts using diverse sampling strategies and whole-genome sequencing.
Keywords: avian faecal droppings; avian influenza; cloacal swabs; wetlands; whole-genome sequencing; wild birds.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures

Similar articles
-
Avian Influenza Viruses in Wild Birds: Virus Evolution in a Multihost Ecosystem.J Virol. 2018 Jul 17;92(15):e00433-18. doi: 10.1128/JVI.00433-18. Print 2018 Aug 1. J Virol. 2018. PMID: 29769347 Free PMC article.
-
Genetic and Pathogenic Characterization of Avian Influenza Virus in Migratory Birds between 2015 and 2019 in Central China.Microbiol Spectr. 2022 Aug 31;10(4):e0165222. doi: 10.1128/spectrum.01652-22. Epub 2022 Jul 12. Microbiol Spectr. 2022. PMID: 35862978 Free PMC article.
-
Epidemiology and molecular characterization of avian influenza A viruses H5N1 and H3N8 subtypes in poultry farms and live bird markets in Bangladesh.Sci Rep. 2023 May 16;13(1):7912. doi: 10.1038/s41598-023-33814-8. Sci Rep. 2023. PMID: 37193732 Free PMC article.
-
Review analysis and impact of co-circulating H5N1 and H9N2 avian influenza viruses in Bangladesh.Epidemiol Infect. 2018 Jul;146(10):1259-1266. doi: 10.1017/S0950268818001292. Epub 2018 May 21. Epidemiol Infect. 2018. PMID: 29781424 Free PMC article.
-
Avian influenza in the Greater Mekong Subregion, 2003-2018.Infect Genet Evol. 2019 Oct;74:103920. doi: 10.1016/j.meegid.2019.103920. Epub 2019 Jun 13. Infect Genet Evol. 2019. PMID: 31201870 Review.
References
-
- Walker P.J., Siddell S.G., Lefkowitz E.J., Mushegian A.R., Adriaenssens E.M., Dempsey D.M., Dutilh B.E., Harrach B., Harrison R.L., Hendrickson R.C., et al. Changes to virus taxonomy and the Statutes ratified by the International Committee on Taxonomy of Viruses (2020) Arch. Virol. 2020;165:2737–2748. doi: 10.1007/s00705-020-04752-x. - DOI - PubMed
-
- Stallknecht D.E., Brown J.D. Ecology of Avian Influenza in Wild Birds. In: Swayne D.E., editor. Avian Influenza. Wiley-Blackwell; Ames, IA, USA: 2008. pp. 43–58.
-
- Bevins S., Shriner S., Cumbee J., Dilione K., Douglass K., Ellis J., Killian M.L., Torchetti M., Lenoch J. Intercontinental Movement of Highly Pathogenic Avian Influenza A(H5N1) Clade 2.3.4.4 Virus to the United States, 2021. Emerg. Infect. Dis. 2022;28:1006. doi: 10.3201/eid2805.220318. - DOI - PMC - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
