

This is a preprint.
Recharacterization of RSL3 reveals that the selenoproteome is a druggable target in colorectal cancer
- PMID: 38617233
- PMCID: PMC11014488
- DOI: 10.1101/2024.03.29.587381
Recharacterization of RSL3 reveals that the selenoproteome is a druggable target in colorectal cancer
Update in
-
Recharacterization of the Tumor Suppressive Mechanism of RSL3 Identifies the Selenoproteome as a Druggable Pathway in Colorectal Cancer.Cancer Res. 2025 Aug 1;85(15):2788-2804. doi: 10.1158/0008-5472.CAN-24-3478. Cancer Res. 2025. PMID: 40392234
Abstract
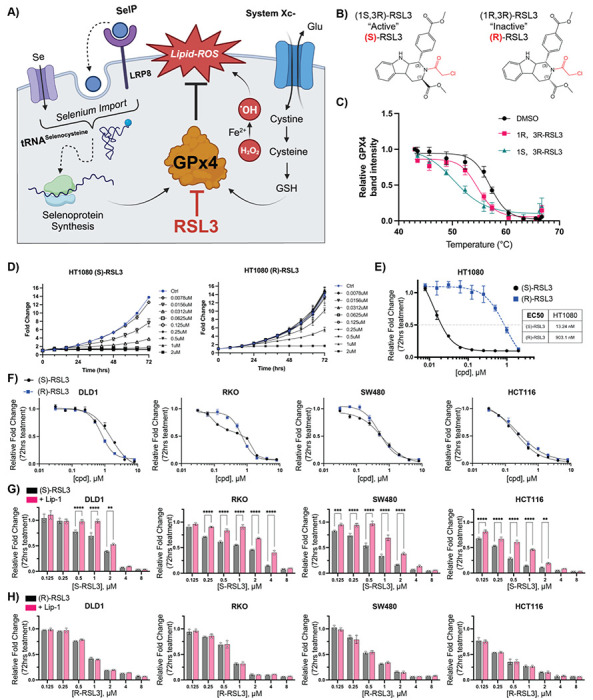
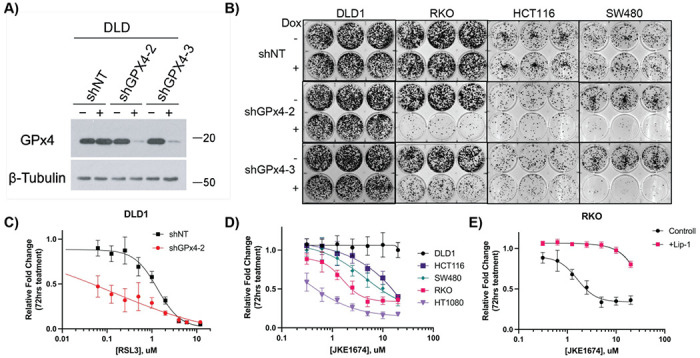
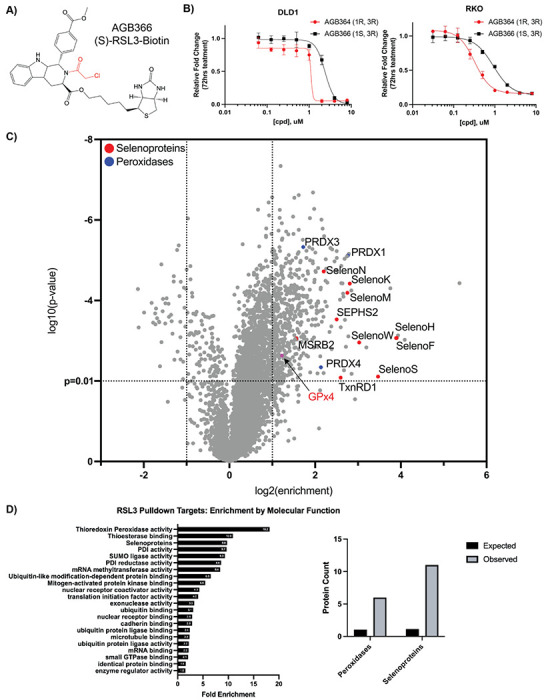
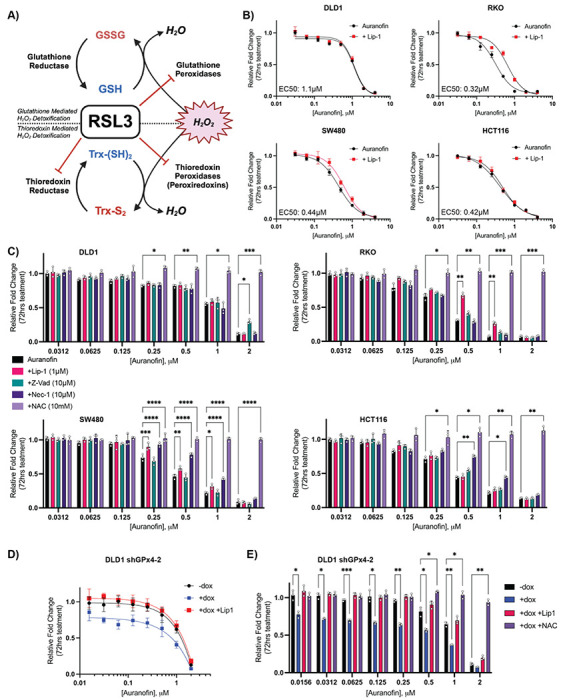
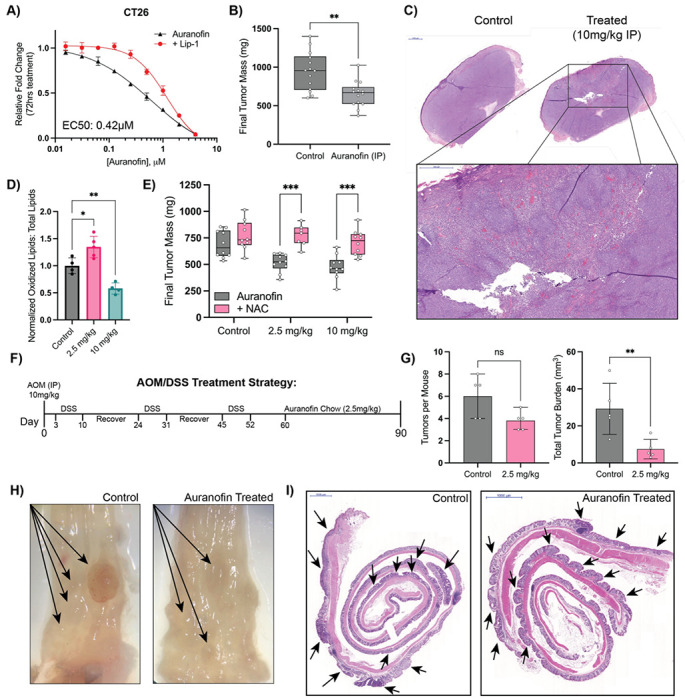
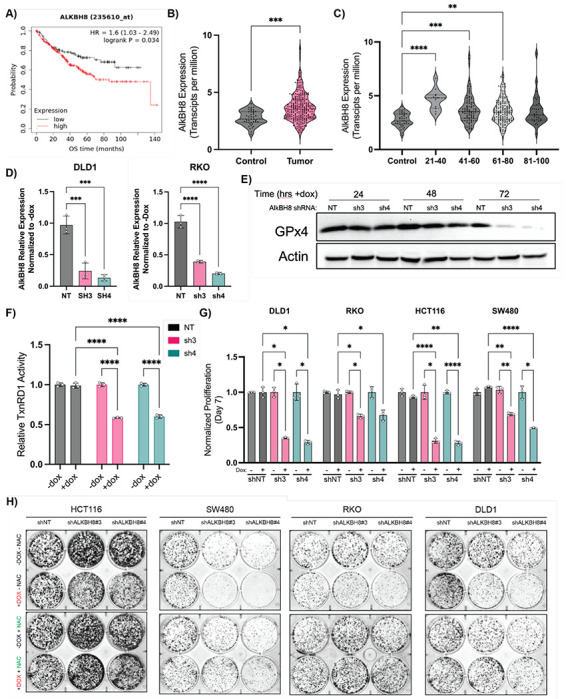
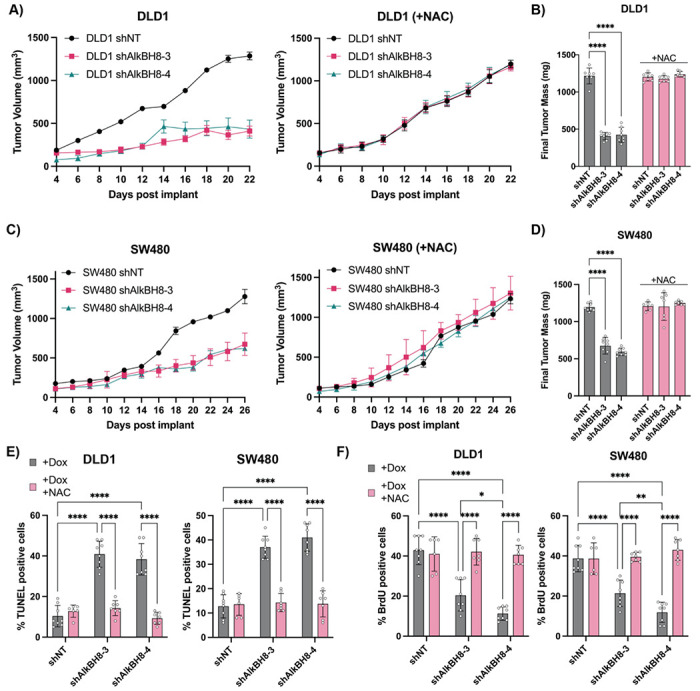
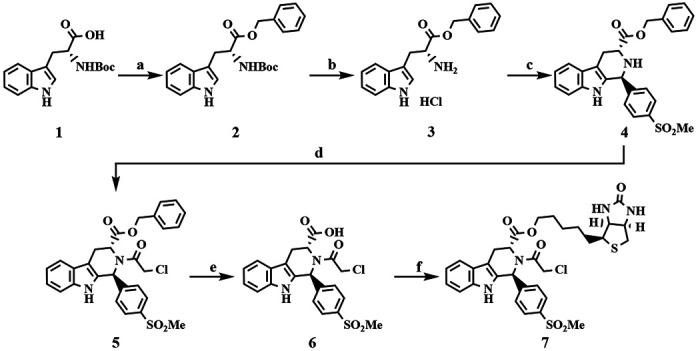
Ferroptosis is a non-apoptotic form of cell death resulting from the iron-dependent accumulation of lipid peroxides. Colorectal cancer (CRC) cells accumulate high levels of intracellular iron and reactive oxygen species (ROS) and are thus particularly sensitive to ferroptosis. The compound (S)-RSL3 ([1S,3R]-RSL3) is a commonly used ferroptosis inducing compound that is currently characterized as a selective inhibitor of the selenocysteine containing enzyme (selenoprotein) Gluathione Peroxidase 4 (GPx4), an enzyme that utilizes glutathione to directly detoxify lipid peroxides. However, through chemical controls utilizing the (R) stereoisomer of RSL3 ([1R,3R]-RSL3) that does not bind GPx4, combined with inducible genetic knockdowns of GPx4 in CRC cell lines, we revealed that GPx4 dependency does not always align with (S)-RSL3 sensitivity, questioning the current characterization of GPx4 as the central regulator of ferroptosis. Utilizing affinity pull-down mass spectrometry with chemically modified (S)-RSL3 probes we discovered that the effects of (S)-RSL3 extend far beyond GPx4 inhibition, revealing that (S)-RSL3 is a broad and non-selective inhibitor of selenoproteins. To further investigate the therapeutic potential of broadly disrupting the selenoproteome as a therapeutic strategy in CRC, we employed additional chemical and genetic approaches. We found that the selenoprotein inhibitor auranofin, an FDA approved gold-salt, chemically induced oxidative cell death and ferroptosis in both in-vitro and in-vivo models of CRC. Consistent with these data, we found that AlkBH8, a tRNA-selenocysteine methyltransferase required for the translation of selenoproteins, is essential for the in-vitro growth and xenograft survival of CRC cell lines. In summary, these findings recharacterize the mechanism of action of the most commonly used ferroptosis inducing molecule, (S)-RSL3, and reveal that broad inhibition of selenoproteins is a promising novel therapeutic angle for the treatment of CRC.
Figures
References
-
- Siegel R. L., Giaquinto A. N. & Jemal A. Cancer statistics, 2024. CA. Cancer J. Clin. 74, 12–49 (2024). - PubMed
-
- Siegel R. L., Wagle N. S., Cercek A., Smith R. A. & Jemal A. Colorectal cancer statistics, 2023. CA. Cancer J. Clin. 73, 233–254 (2023). - PubMed
-
- Raskov H., Søby J. H., Troelsen J., Bojesen R. D. & Gögenur I. Driver Gene Mutations and Epigenetics in Colorectal Cancer. Ann. Surg. 271, 75 (2020). - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources