

Evaluation of the Patients with the Diagnosis of Pontocerebellar Hypoplasia: A Multicenter National Study
- PMID: 38622473
- PMCID: PMC11489189
- DOI: 10.1007/s12311-024-01690-1
Evaluation of the Patients with the Diagnosis of Pontocerebellar Hypoplasia: A Multicenter National Study
Abstract
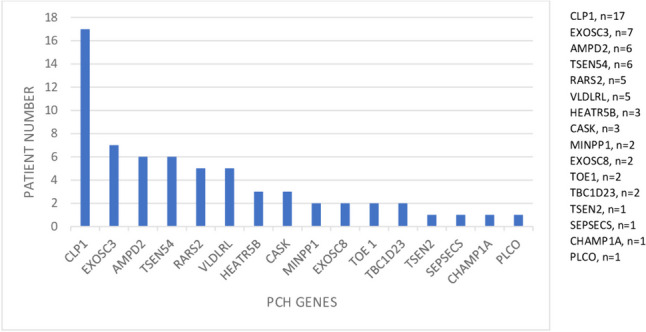
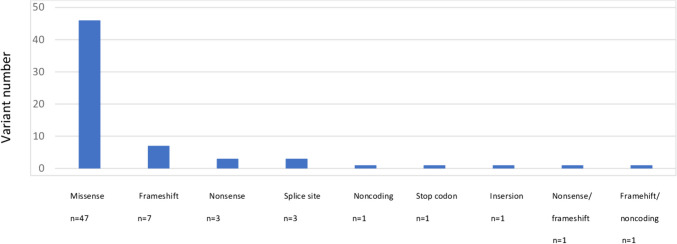
Pontocerebellar hypoplasia (PCH) is a heterogeneous group of neurodegenerative disorders characterized by hypoplasia and degeneration of the cerebellum and pons. We aimed to identify the clinical, laboratory, and imaging findings of the patients with diagnosed PCH with confirmed genetic analysis. We collected available clinical data, laboratory, and imaging findings in our retrospective multicenter national study of 64 patients with PCH in Turkey. The genetic analysis included the whole-exome sequencing (WES), targeted next-generation sequencing (NGS), or single gene analysis. Sixty-four patients with PCH were 28 female (43.8%) and 36 (56.3%) male. The patients revealed homozygous mutation in 89.1%, consanguinity in 79.7%, pregnancy at term in 85.2%, microcephaly in 91.3%, psychomotor retardation in 98.4%, abnormal neurological findings in 100%, seizure in 63.8%, normal biochemistry and metabolic investigations in 92.2%, and dysmorphic findings in 51.2%. The missense mutation was found to be the most common variant type in all patients with PCH. It was detected as CLP1 (n = 17) was the most common PCH related gene. The homozygous missense variant c.419G > A (p.Arg140His) was identified in all patients with CLP1. Moreover, all patients showed the same homozygous missense variant c.919G > T (p.A307S) in TSEN54 group (n = 6). In Turkey, CLP1 was identified as the most common causative gene with the identical variant c.419G > A; p.Arg140His. The current study supports that genotype data on PCH leads to phenotypic variability over a wide phenotypic spectrum.
Keywords: CLP1; Genotype; Phenotype; Pontocerebellar Hypoplasia.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
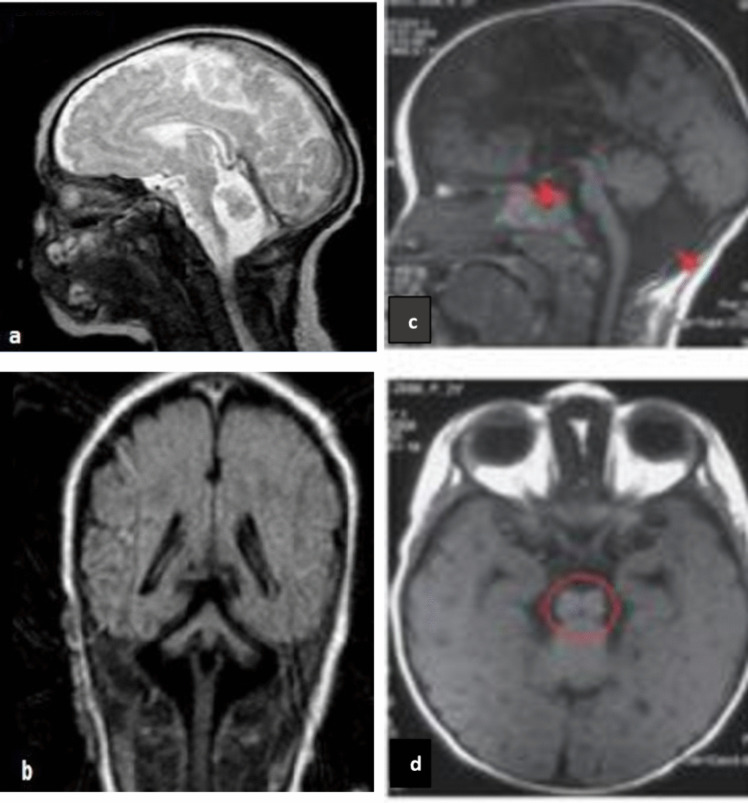
Figures
References
-
- Nuovo S, Micalizzi A, Romaniello R, Arrigoni F, Ginevrino M, Casella A, et al. Refining the mutational spectrum and gene-phenotype correlates in pontocerebellar hypoplasia: results of a multicentric study. J Med Genet. 2022;59(4):399–409. - PubMed
-
- Brun R. Zur Kenntnis der Bildungsfehler des Kleinhirns. I. Über Aplasie und Hypoplasie des Neozerebellums. Schweiz Arch Neurol Psychiat. 1917;1:61–123.
-
- Barth PG, Vrensen GF, Uylings HB, Oorthuys JW, Stam FC. Inherited syndrome of microcephaly, dyskinesia and pontocerebellar hypoplasia: a systemic atrophy with early onset. J Neurol Sci. 1990;97(1):25–42. - PubMed
Publication types
MeSH terms
Supplementary concepts
LinkOut - more resources
Full Text Sources
Medical
