

CAG repeat mosaicism is gene specific in spinocerebellar ataxias
- PMID: 38626762
- PMCID: PMC11080609
- DOI: 10.1016/j.ajhg.2024.03.015
CAG repeat mosaicism is gene specific in spinocerebellar ataxias
Abstract
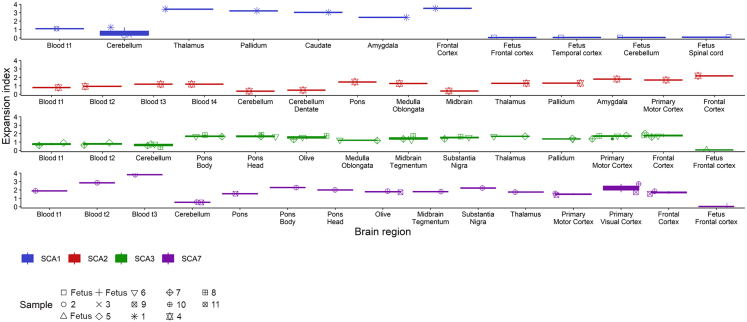
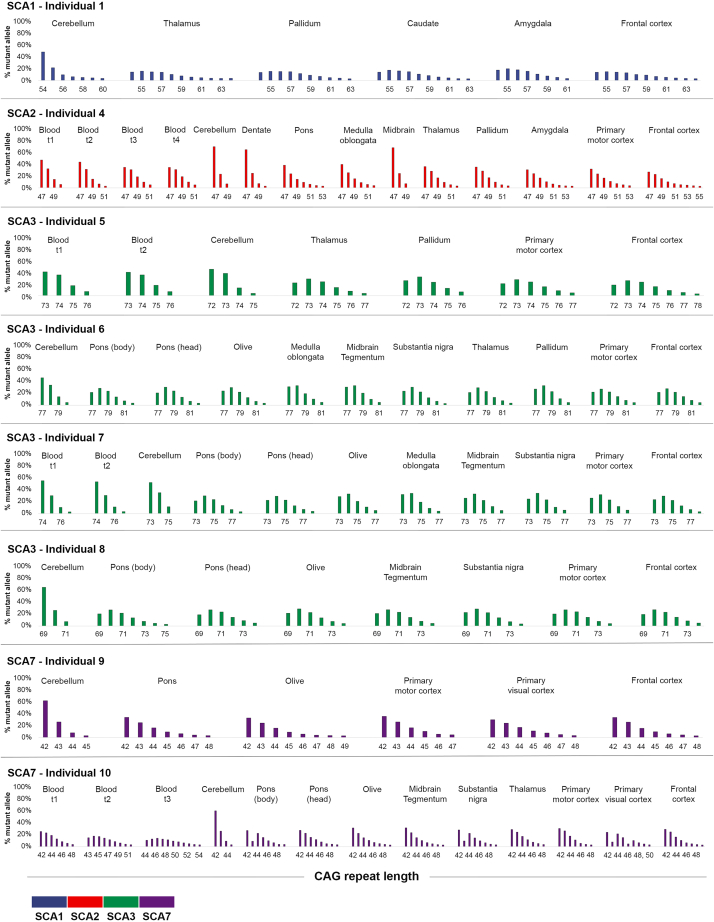
Expanded CAG repeats in coding regions of different genes are the most common cause of dominantly inherited spinocerebellar ataxias (SCAs). These repeats are unstable through the germline, and larger repeats lead to earlier onset. We measured somatic expansion in blood samples collected from 30 SCA1, 50 SCA2, 74 SCA3, and 30 SCA7 individuals over a mean interval of 8.5 years, along with postmortem tissues and fetal tissues from SCA1, SCA3, and SCA7 individuals to examine somatic expansion at different stages of life. We showed that somatic mosaicism in the blood increases over time. Expansion levels are significantly different among SCAs and correlate with CAG repeat lengths. The level of expansion is greater in individuals with SCA7 who manifest disease compared to that of those who do not yet display symptoms. Brain tissues from SCA individuals have larger expansions compared to the blood. The cerebellum has the lowest mosaicism among the studied brain regions, along with a high expression of ATXNs and DNA repair genes. This was the opposite in cortices, with the highest mosaicism and lower expression of ATXNs and DNA repair genes. Fetal cortices did not show repeat instability. This study shows that CAG repeats are increasingly unstable during life in the blood and the brain of SCA individuals, with gene- and tissue-specific patterns.
Keywords: ATXN1; ATXN2; ATXN3; ATXN7; CAG repeat; DNA repair; SCA; repeat expansion; somatic instability; spinocerebellar ataxia.
Copyright © 2024 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests The authors declare no competing interests.
Figures




References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
