

Differentiation shifts from a reversible to an irreversible heterochromatin state at the DM1 locus
- PMID: 38627364
- PMCID: PMC11021500
- DOI: 10.1038/s41467-024-47217-4
Differentiation shifts from a reversible to an irreversible heterochromatin state at the DM1 locus
Abstract
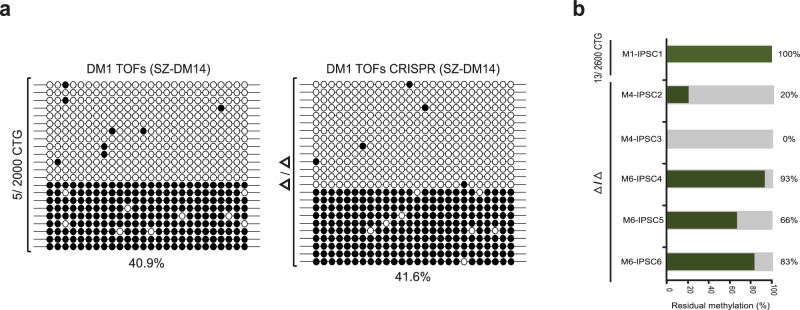
Epigenetic defects caused by hereditary or de novo mutations are implicated in various human diseases. It remains uncertain whether correcting the underlying mutation can reverse these defects in patient cells. Here we show by the analysis of myotonic dystrophy type 1 (DM1)-related locus that in mutant human embryonic stem cells (hESCs), DNA methylation and H3K9me3 enrichments are completely abolished by repeat excision (CTG2000 expansion), whereas in patient myoblasts (CTG2600 expansion), repeat deletion fails to do so. This distinction between undifferentiated and differentiated cells arises during cell differentiation, and can be reversed by reprogramming of gene-edited myoblasts. We demonstrate that abnormal methylation in DM1 is distinctively maintained in the undifferentiated state by the activity of the de novo DNMTs (DNMT3b in tandem with DNMT3a). Overall, the findings highlight a crucial difference in heterochromatin maintenance between undifferentiated (sequence-dependent) and differentiated (sequence-independent) cells, thus underscoring the role of differentiation as a locking mechanism for repressive epigenetic modifications at the DM1 locus.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures




References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
