

Deep learning methods in metagenomics: a review
- PMID: 38630611
- PMCID: PMC11092122
- DOI: 10.1099/mgen.0.001231
Deep learning methods in metagenomics: a review
Abstract
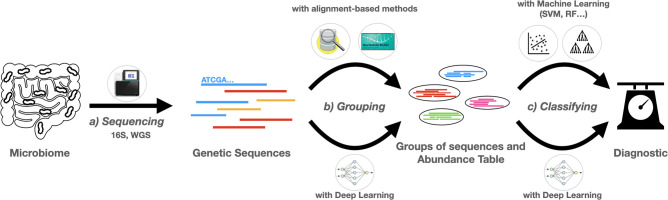
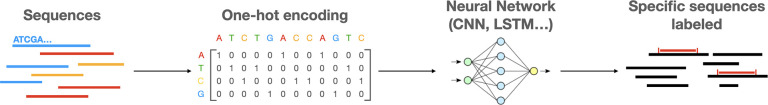
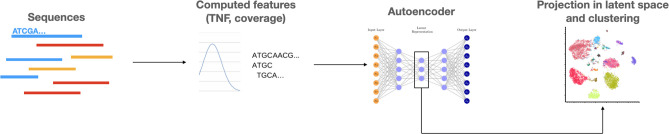
The ever-decreasing cost of sequencing and the growing potential applications of metagenomics have led to an unprecedented surge in data generation. One of the most prevalent applications of metagenomics is the study of microbial environments, such as the human gut. The gut microbiome plays a crucial role in human health, providing vital information for patient diagnosis and prognosis. However, analysing metagenomic data remains challenging due to several factors, including reference catalogues, sparsity and compositionality. Deep learning (DL) enables novel and promising approaches that complement state-of-the-art microbiome pipelines. DL-based methods can address almost all aspects of microbiome analysis, including novel pathogen detection, sequence classification, patient stratification and disease prediction. Beyond generating predictive models, a key aspect of these methods is also their interpretability. This article reviews DL approaches in metagenomics, including convolutional networks, autoencoders and attention-based models. These methods aggregate contextualized data and pave the way for improved patient care and a better understanding of the microbiome's key role in our health.
Keywords: binning; deep learning; disease prediction; embedding; metagenomics; microbiome; neural network.
Conflict of interest statement
The authors declare no competing interests.
Figures
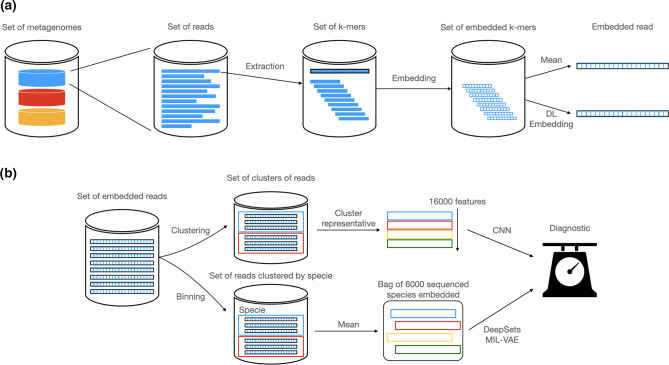
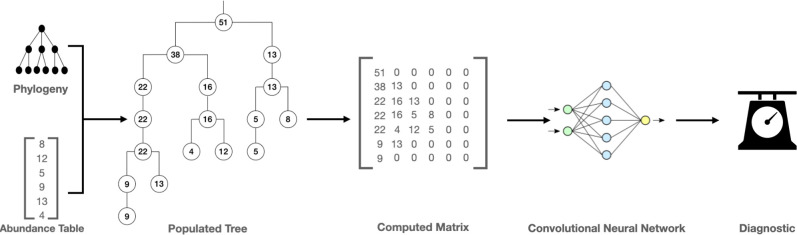
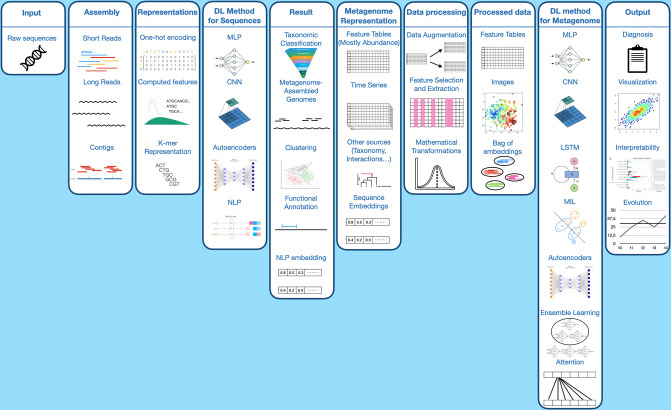
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources