

Probabilistic association of differentially expressed genes with cis-regulatory elements
- PMID: 38631728
- PMCID: PMC11146588
- DOI: 10.1101/gr.278598.123
Probabilistic association of differentially expressed genes with cis-regulatory elements
Abstract
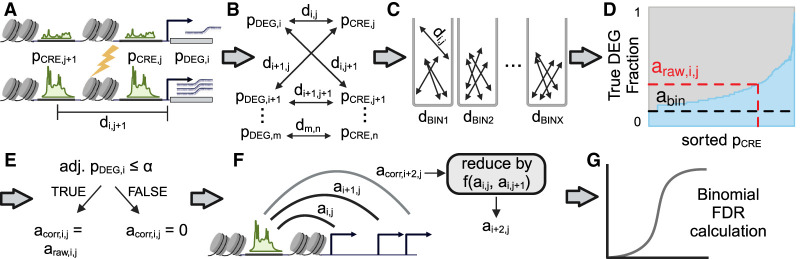
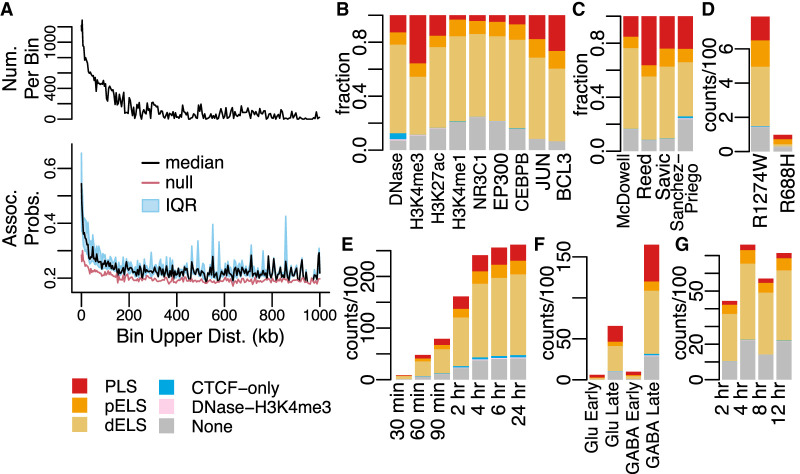
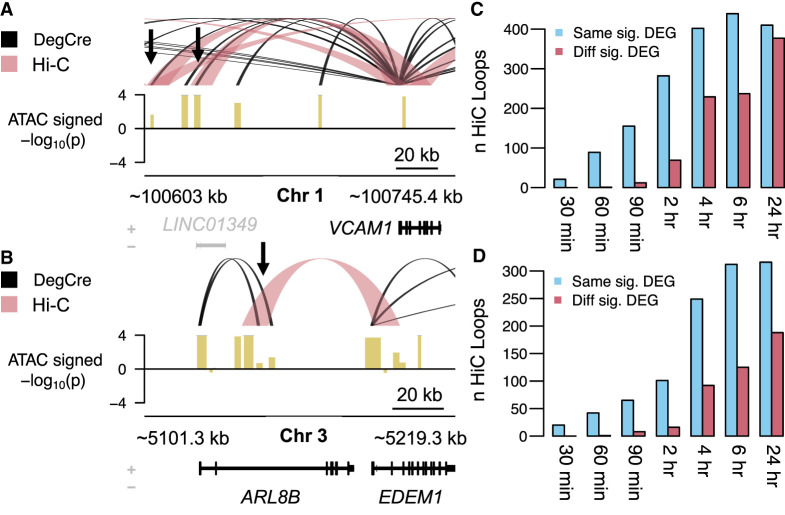
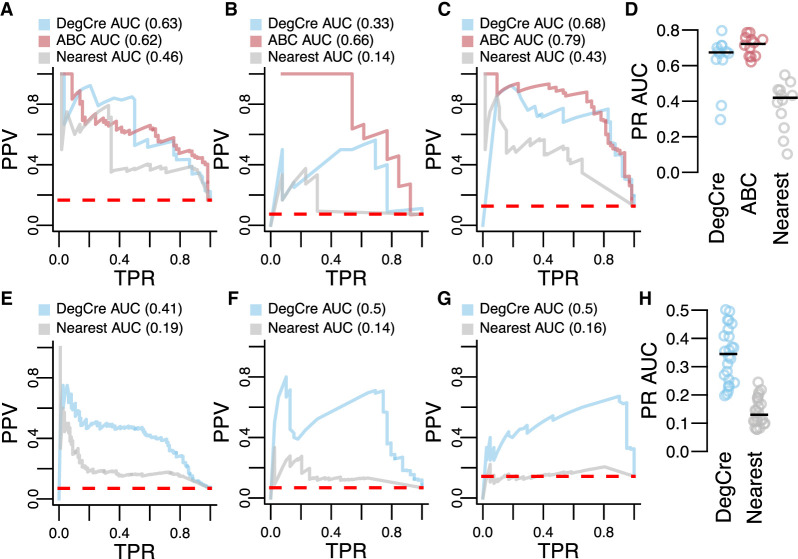
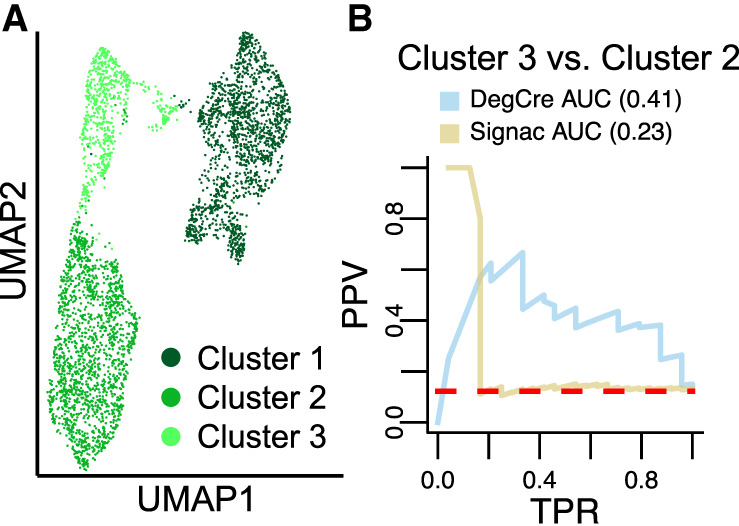
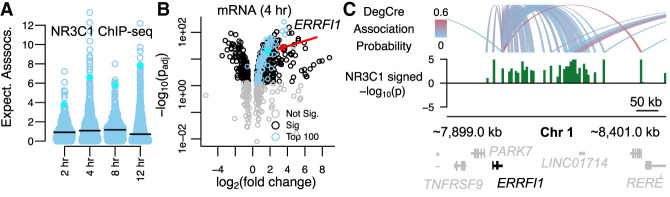
Differential gene expression in response to perturbations is mediated at least in part by changes in binding of transcription factors (TFs) and other proteins at specific genomic regions. Association of these cis-regulatory elements (CREs) with their target genes is a challenging task that is essential to address many biological and mechanistic questions. Many current approaches rely on chromatin conformation capture techniques or single-cell correlational methods to establish CRE-to-gene associations. These methods can be effective but have limitations, including resolution, gaps in detectable association distances, and cost. As an alternative, we have developed DegCre, a nonparametric method that evaluates correlations between measurements of perturbation-induced differential gene expression and differential regulatory signal at CREs to score possible CRE-to-gene associations. It has several unique features, including the ability to use any type of CRE activity measurement, yield probabilistic scores for CRE-to-gene pairs, and assess CRE-to-gene pairings across a wide range of sequence distances. We apply DegCre to six data sets, each using different perturbations and containing a variety of regulatory signal measurements, including chromatin openness, histone modifications, and TF occupancy. To test their efficacy, we compare DegCre associations to Hi-C loop calls and CRISPR-validated CRE-to-gene associations, establishing good performance by DegCre that is comparable or superior to competing methods. DegCre is a novel approach to the association of CREs to genes from a perturbation-differential perspective, with strengths that are complementary to existing approaches and allow for new insights into gene regulation.
© 2024 Roberts et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
Similar articles
-
De novo prediction of cis-regulatory elements and modules through integrative analysis of a large number of ChIP datasets.BMC Genomics. 2014 Dec 2;15:1047. doi: 10.1186/1471-2164-15-1047. BMC Genomics. 2014. PMID: 25442502 Free PMC article.
-
Multiscale footprints reveal the organization of cis-regulatory elements.Nature. 2025 Feb;638(8051):779-786. doi: 10.1038/s41586-024-08443-4. Epub 2025 Jan 22. Nature. 2025. PMID: 39843737 Free PMC article.
-
CREATE: cell-type-specific cis-regulatory element identification via discrete embedding.Nat Commun. 2025 May 17;16(1):4607. doi: 10.1038/s41467-025-59780-5. Nat Commun. 2025. PMID: 40382355 Free PMC article.
-
Chromatin modules and their implication in genomic organization and gene regulation.Trends Genet. 2023 Feb;39(2):140-153. doi: 10.1016/j.tig.2022.11.003. Epub 2022 Dec 20. Trends Genet. 2023. PMID: 36549923 Review.
-
Shaping gene expression and its evolution by chromatin architecture and enhancer activity.Curr Top Dev Biol. 2024;159:406-437. doi: 10.1016/bs.ctdb.2024.01.001. Epub 2024 Feb 1. Curr Top Dev Biol. 2024. PMID: 38729683 Review.
Cited by
-
Non-coding variation in dementias: mechanisms, insights, and challenges.NPJ Dement. 2025;1(1):9. doi: 10.1038/s44400-025-00012-4. Epub 2025 Jun 3. NPJ Dement. 2025. PMID: 40476256 Free PMC article. Review.
-
Analyzing super-enhancer temporal dynamics reveals potential critical enhancers and their gene regulatory networks underlying skeletal muscle development.Genome Res. 2024 Dec 23;34(12):2190-2202. doi: 10.1101/gr.278344.123. Genome Res. 2024. PMID: 39433439 Free PMC article.
References
-
- Adamson B, Norman TM, Jost M, Cho MY, Nuñez JK, Chen Y, Villalta JE, Gilbert LA, Horlbeck MA, Hein MY, et al. 2016. A multiplexed single-cell CRISPR screening platform enables systematic dissection of the unfolded protein response. Cell 167: 1867–1882.e21. 10.1016/j.cell.2016.11.048 - DOI - PMC - PubMed
-
- Bardy C, van den Hurk M, Eames T, Marchand C, Hernandez RV, Kellogg M, Gorris M, Galet B, Palomares V, Brown J, et al. 2015. Neuronal medium that supports basic synaptic functions and activity of human neurons in vitro. Proc Natl Acad Sci USA 112: E2725–E2734. 10.1073/pnas.1504393112 - DOI - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous