

Type I interferon regulation by USP18 is a key vulnerability in cancer
- PMID: 38632987
- PMCID: PMC11022047
- DOI: 10.1016/j.isci.2024.109593
Type I interferon regulation by USP18 is a key vulnerability in cancer
Abstract
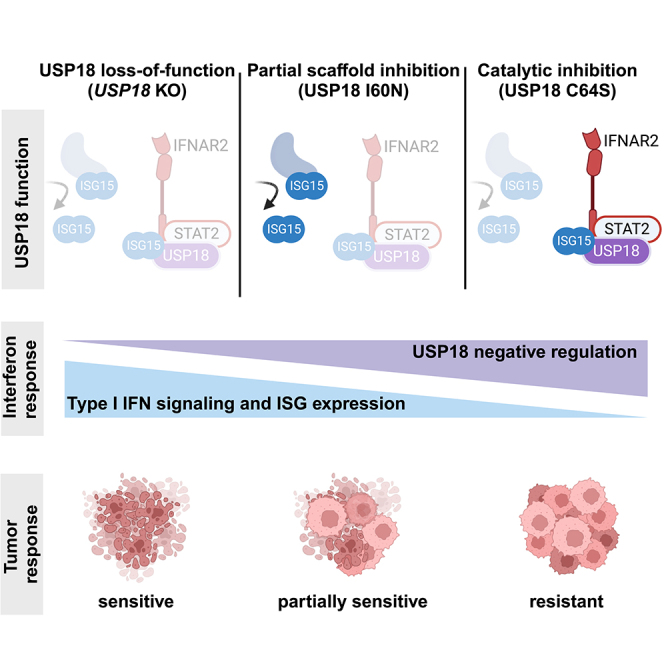
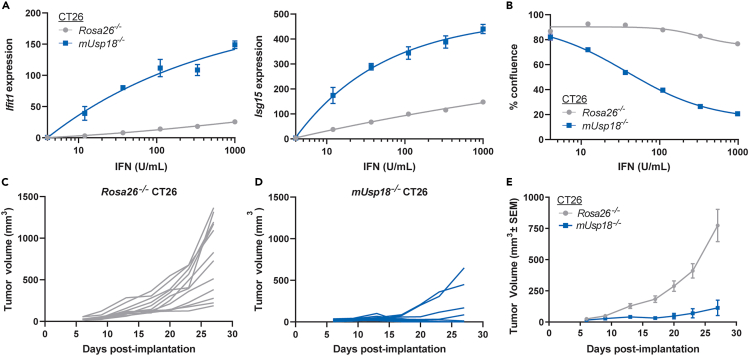
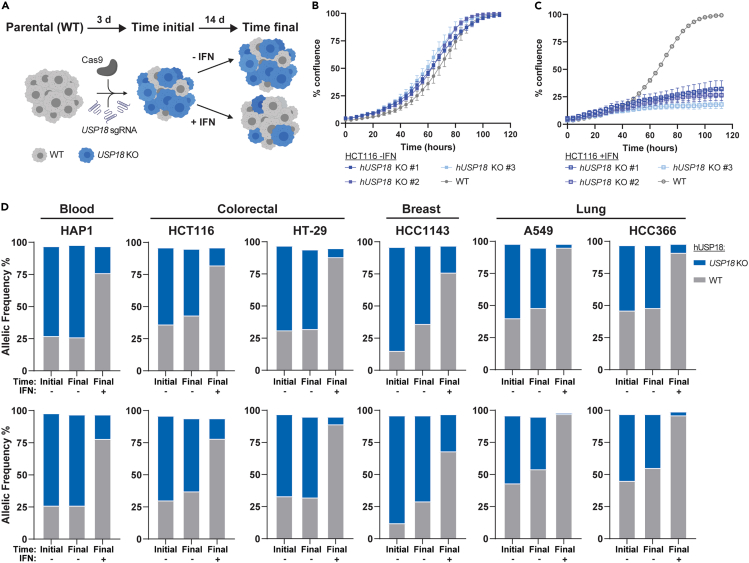
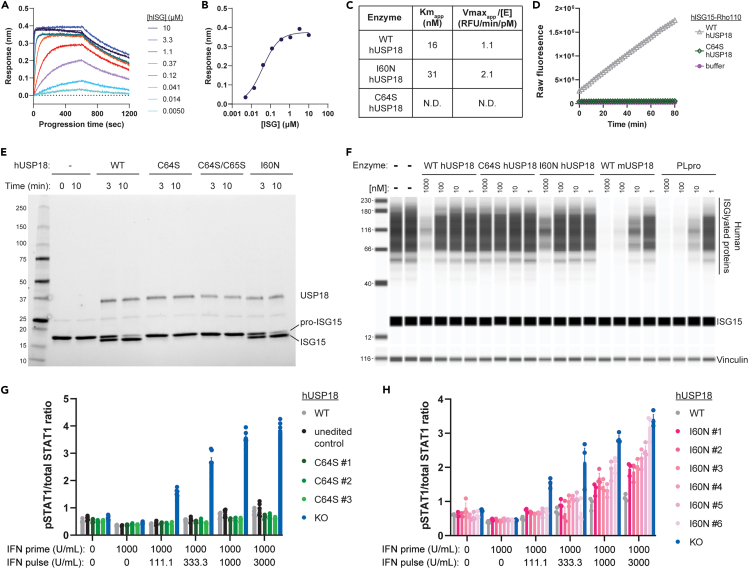
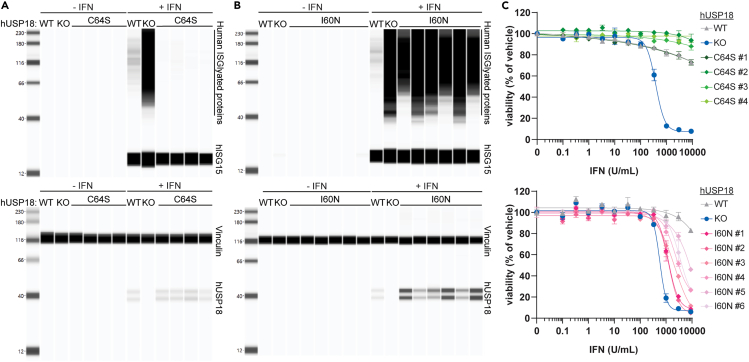
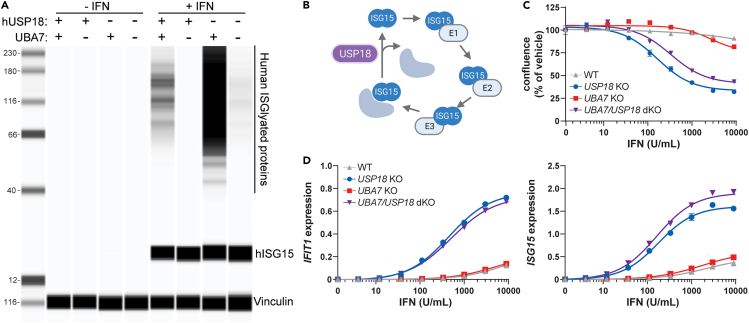
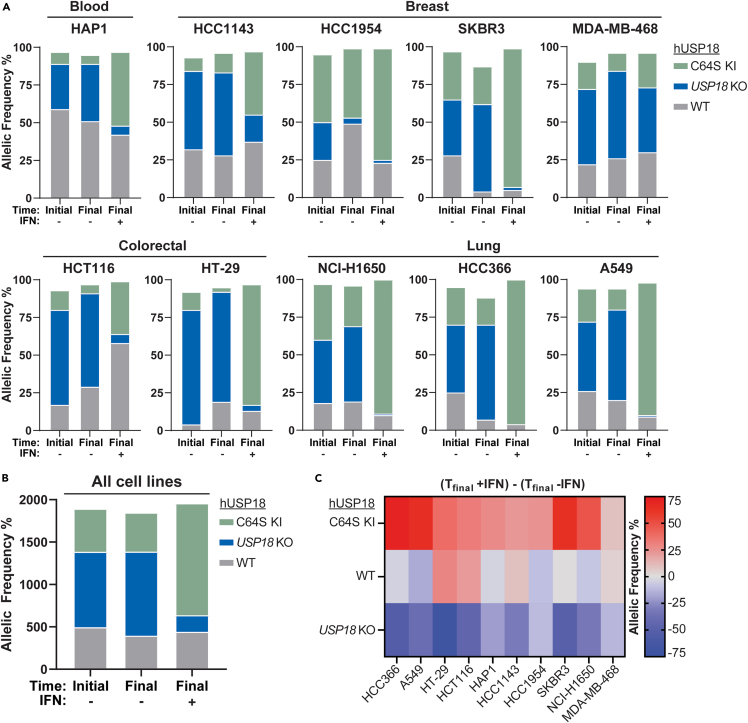
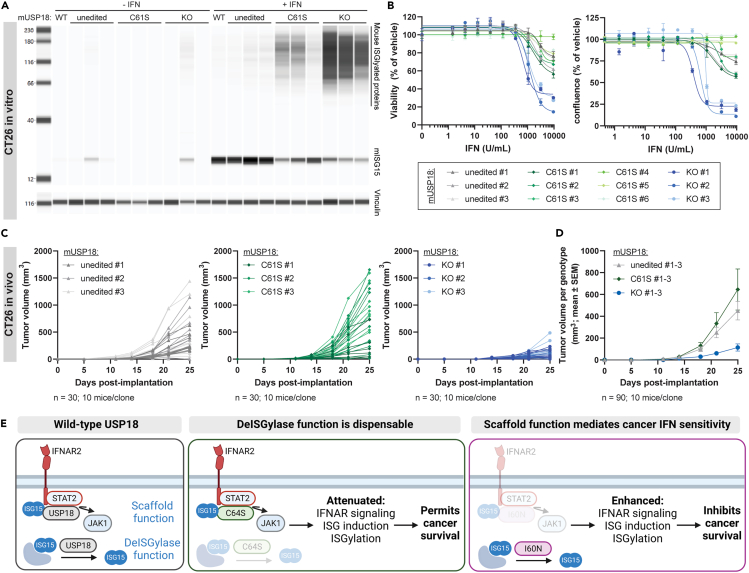
Precise regulation of Type I interferon signaling is crucial for combating infection and cancer while avoiding autoimmunity. Type I interferon signaling is negatively regulated by USP18. USP18 cleaves ISG15, an interferon-induced ubiquitin-like modification, via its canonical catalytic function, and inhibits Type I interferon receptor activity through its scaffold role. USP18 loss-of-function dramatically impacts immune regulation, pathogen susceptibility, and tumor growth. However, prior studies have reached conflicting conclusions regarding the relative importance of catalytic versus scaffold function. Here, we develop biochemical and cellular methods to systematically define the physiological role of USP18. By comparing a patient-derived mutation impairing scaffold function (I60N) to a mutation disrupting catalytic activity (C64S), we demonstrate that scaffold function is critical for cancer cell vulnerability to Type I interferon. Surprisingly, we discovered that human USP18 exhibits minimal catalytic activity, in stark contrast to mouse USP18. These findings resolve human USP18's mechanism-of-action and enable USP18-targeted therapeutics.
Keywords: Cancer; Cell biology; Immune response; Immunity.
© 2024 Pfizer Inc.
Conflict of interest statement
VJ, HW, CWL, DRH, KL, ECR, MY, ZKJ, EC, YX, JS, BY, AG, PL, ACG, VF, PDW, and FW are current or former employees of Pfizer and may own Pfizer stock. BMK and APF receive research funding from Pfizer. This work was supported by Pfizer, and APF and BMK are funded by the Chinese Academy of Medical Sciences (CAMS) Innovation Fund for Medical Science (CIFMS), China (grant nr - 2018-I2M-2-002).
Figures
References
LinkOut - more resources
Full Text Sources
Miscellaneous
