

IMPROVE: a feature model to predict neoepitope immunogenicity through broad-scale validation of T-cell recognition
- PMID: 38633261
- PMCID: PMC11021644
- DOI: 10.3389/fimmu.2024.1360281
IMPROVE: a feature model to predict neoepitope immunogenicity through broad-scale validation of T-cell recognition
Abstract
Background: Mutation-derived neoantigens are critical targets for tumor rejection in cancer immunotherapy, and better tools for neoepitope identification and prediction are needed to improve neoepitope targeting strategies. Computational tools have enabled the identification of patient-specific neoantigen candidates from sequencing data, but limited data availability has hindered their capacity to predict which of the many neoepitopes will most likely give rise to T cell recognition.
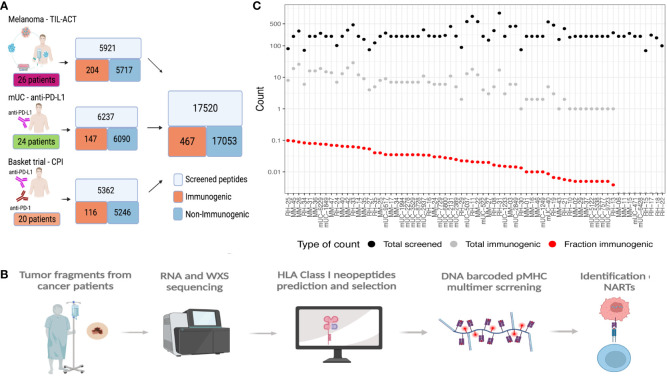
Method: To address this, we make use of experimentally validated T cell recognition towards 17,500 neoepitope candidates, with 467 being T cell recognized, across 70 cancer patients undergoing immunotherapy.
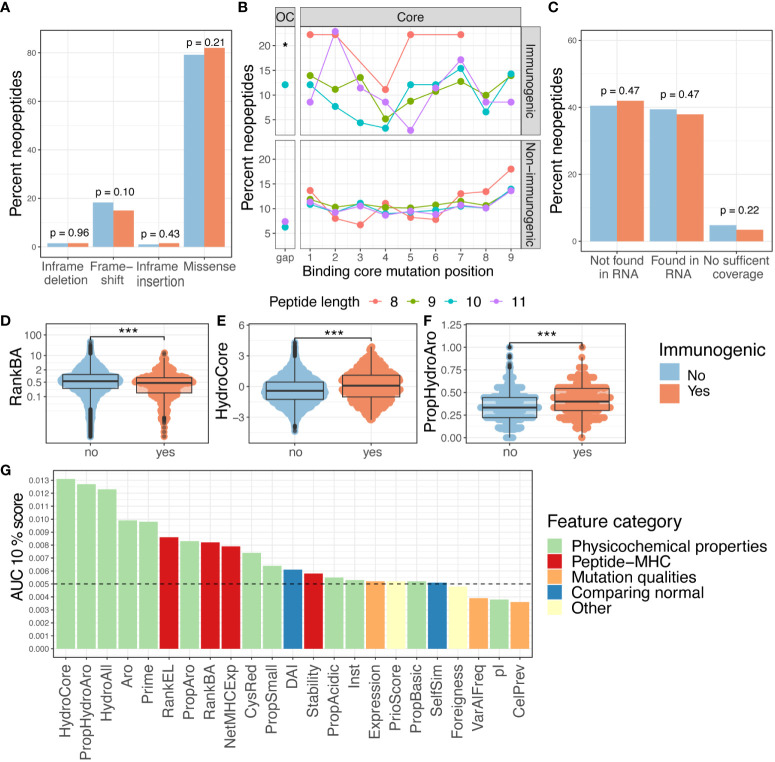
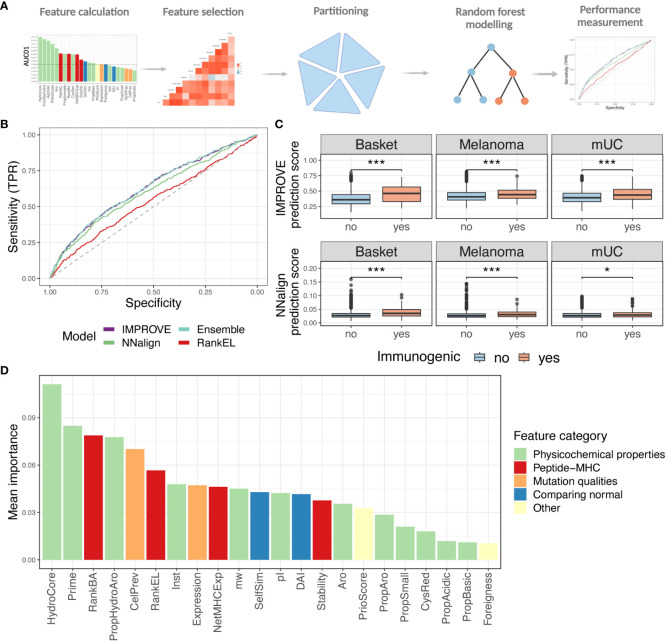
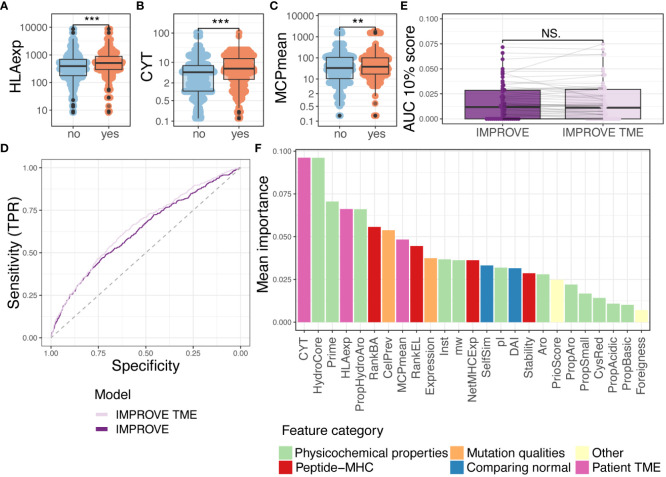
Results: We evaluated 27 neoepitope characteristics, and created a random forest model, IMPROVE, to predict neoepitope immunogenicity. The presence of hydrophobic and aromatic residues in the peptide binding core were the most important features for predicting neoepitope immunogenicity.
Conclusion: Overall, IMPROVE was found to significantly advance the identification of neoepitopes compared to other current methods.
Keywords: immunoinformatics; immunotherapy; machine learning; neoantigen; neoepitope prediction.
Copyright © 2024 Borch, Carri, Reynisson, Alvarez, Munk, Montemurro, Kristensen, Tvingsholm, Holm, Heeke, Moss, Hansen, Schaap-Johansen, Bagger, de Lima, Rohrberg, Funt, Donia, Svane, Lassen, Barra, Nielsen and Hadrup.
Conflict of interest statement
SH is the cofounder of PokeAcell and is the inventor of several licensed patents, however, none of these activities are of relevance to the work presented in this manuscript. The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
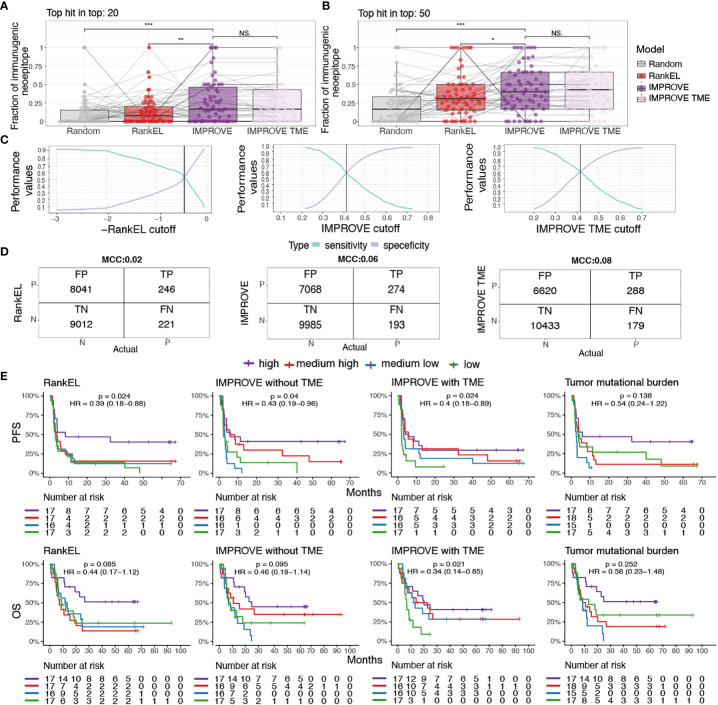
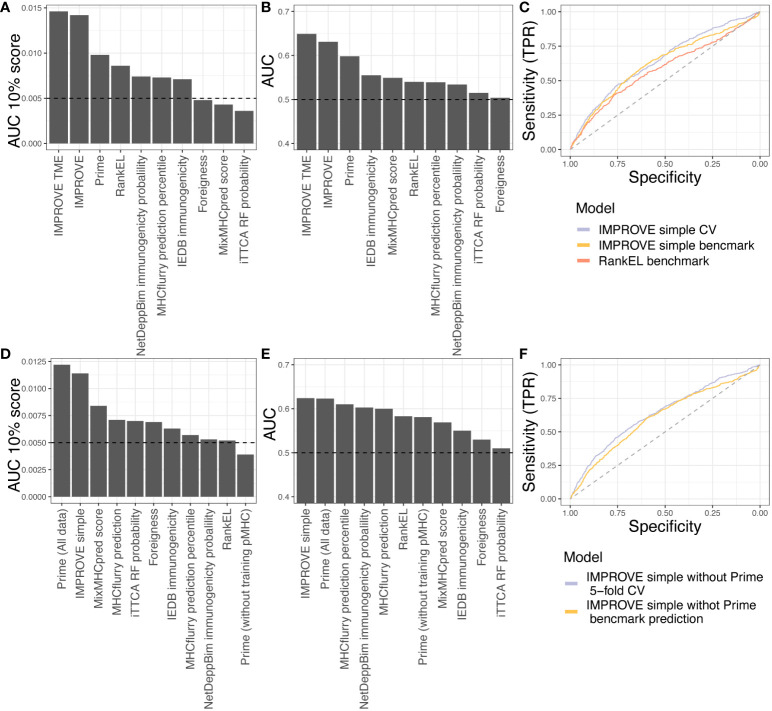
References
-
- Goodman AM, Kato S, Bazhenova L, Patel SP, Frampton GM, Miller V, et al. Companion diagnostic, pharmacogenomic, and cancer biomarkers tumor mutational burden as an independent predictor of response to immunotherapy in diverse cancers. Mol Cancer Ther. (2017) 16(11):2598–608. doi: 10.1158/1535-7163.MCT-17-0386 - DOI - PMC - PubMed
-
- Cristescu R, Aurora-Garg D, Albright A, Xu L, Liu XQ, Loboda A, et al. Tumor mutational burden predicts the efficacy of pembrolizumab monotherapy: A pan-tumor retrospective analysis of participants with advanced solid tumors. J Immunother Cancer. (2022) 10(1). doi: 10.1136/jitc-2021-003091 - DOI - PMC - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases