

Donor whole blood DNA methylation is not a strong predictor of acute graft versus host disease in unrelated donor allogeneic haematopoietic cell transplantation
- PMID: 38633407
- PMCID: PMC11021570
- DOI: 10.3389/fgene.2024.1242636
Donor whole blood DNA methylation is not a strong predictor of acute graft versus host disease in unrelated donor allogeneic haematopoietic cell transplantation
Abstract
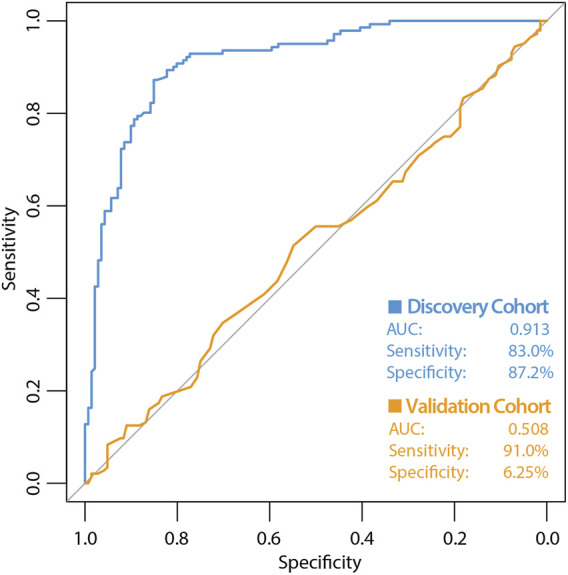
Allogeneic hematopoietic cell transplantation (HCT) is used to treat many blood-based disorders and malignancies, however it can also result in serious adverse events, such as the development of acute graft-versus-host disease (aGVHD). This study aimed to develop a donor-specific epigenetic classifier to reduce incidence of aGVHD by improving donor selection. Genome-wide DNA methylation was assessed in a discovery cohort of 288 HCT donors selected based on recipient aGVHD outcome; this cohort consisted of 144 cases with aGVHD grades III-IV and 144 controls with no aGVHD. We applied a machine learning algorithm to identify CpG sites predictive of aGVHD. Receiver operating characteristic (ROC) curve analysis of these sites resulted in a classifier with an encouraging area under the ROC curve (AUC) of 0.91. To test this classifier, we used an independent validation cohort (n = 288) selected using the same criteria as the discovery cohort. Attempts to validate the classifier failed with the AUC falling to 0.51. These results indicate that donor DNA methylation may not be a suitable predictor of aGVHD in an HCT setting involving unrelated donors, despite the initial promising results in the discovery cohort. Our work highlights the importance of independent validation of machine learning classifiers, particularly when developing classifiers intended for clinical use.
Keywords: DNA methylation; HCT (hematopoietic cell transplant); biomarker identification and validation; epigenetics; haematopoietic stem cell transplant; machine learning.
Copyright © 2024 Webster, Ecker, Moghul, Liu, Dhami, Marzi, Paul, Kuxhausen, Lee, Spellman, Wang, Feber, Rakyan, Peggs and Beck.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures



References
-
- Al-Kadhimi Z., Gul Z., Chen W., Smith D., Abidi M., Deol A., et al. (2014). High incidence of severe acute graft-versus-host disease with tacrolimus and mycophenolate mofetil in a large cohort of related and unrelated allogeneic transplantation patients. Biol. Blood Marrow Transpl. 20, 979–985. 10.1016/j.bbmt.2014.03.016 - DOI - PMC - PubMed
-
- Breiman L. (2001). Random forests. Mach. Learn. 45, 5–32. 10.1023/a:1010933404324 - DOI
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases