

LXR/CD38 activation drives cholesterol-induced macrophage senescence and neurodegeneration via NAD+ depletion
- PMID: 38636518
- PMCID: PMC11223747
- DOI: 10.1016/j.celrep.2024.114102
LXR/CD38 activation drives cholesterol-induced macrophage senescence and neurodegeneration via NAD+ depletion
Abstract
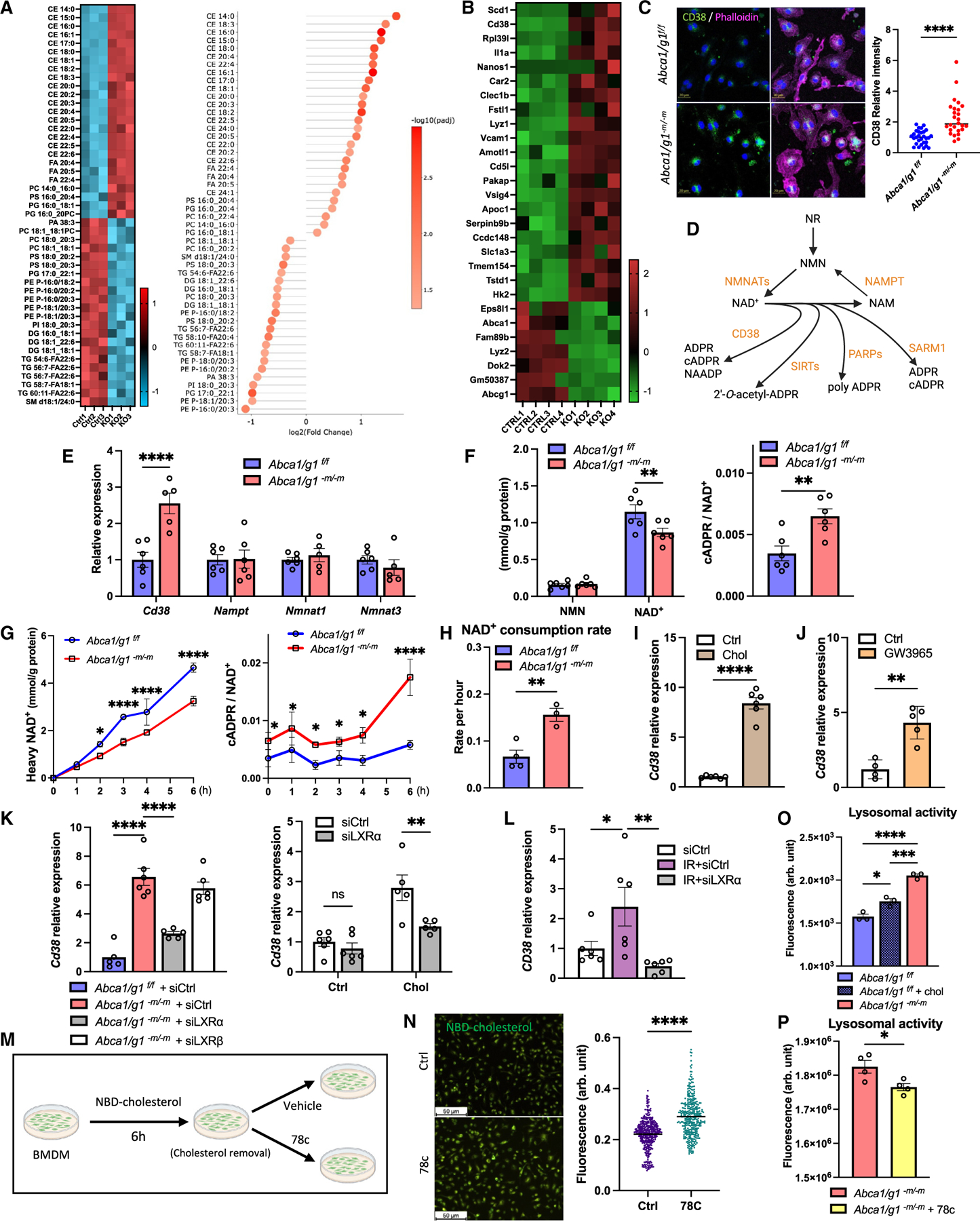
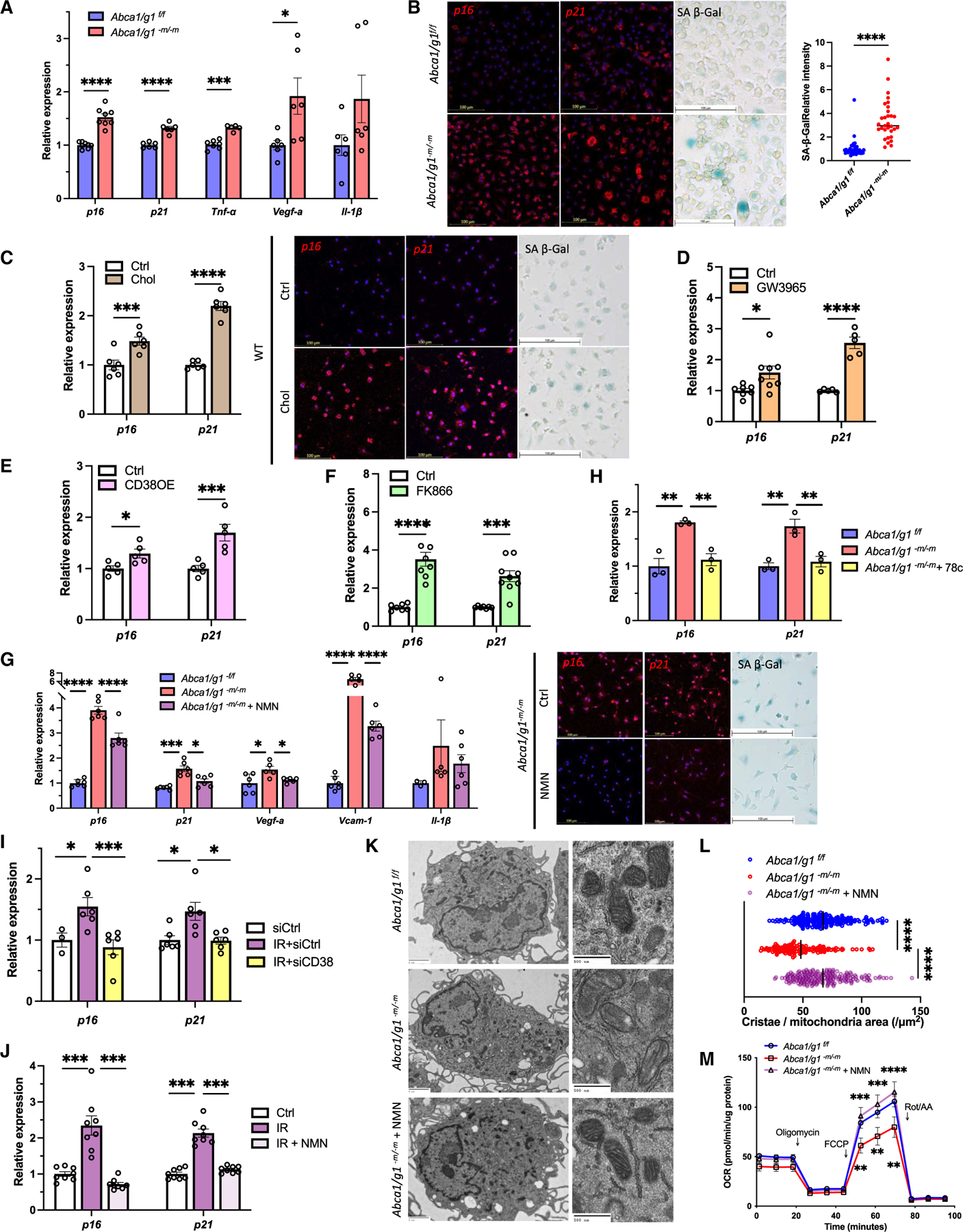
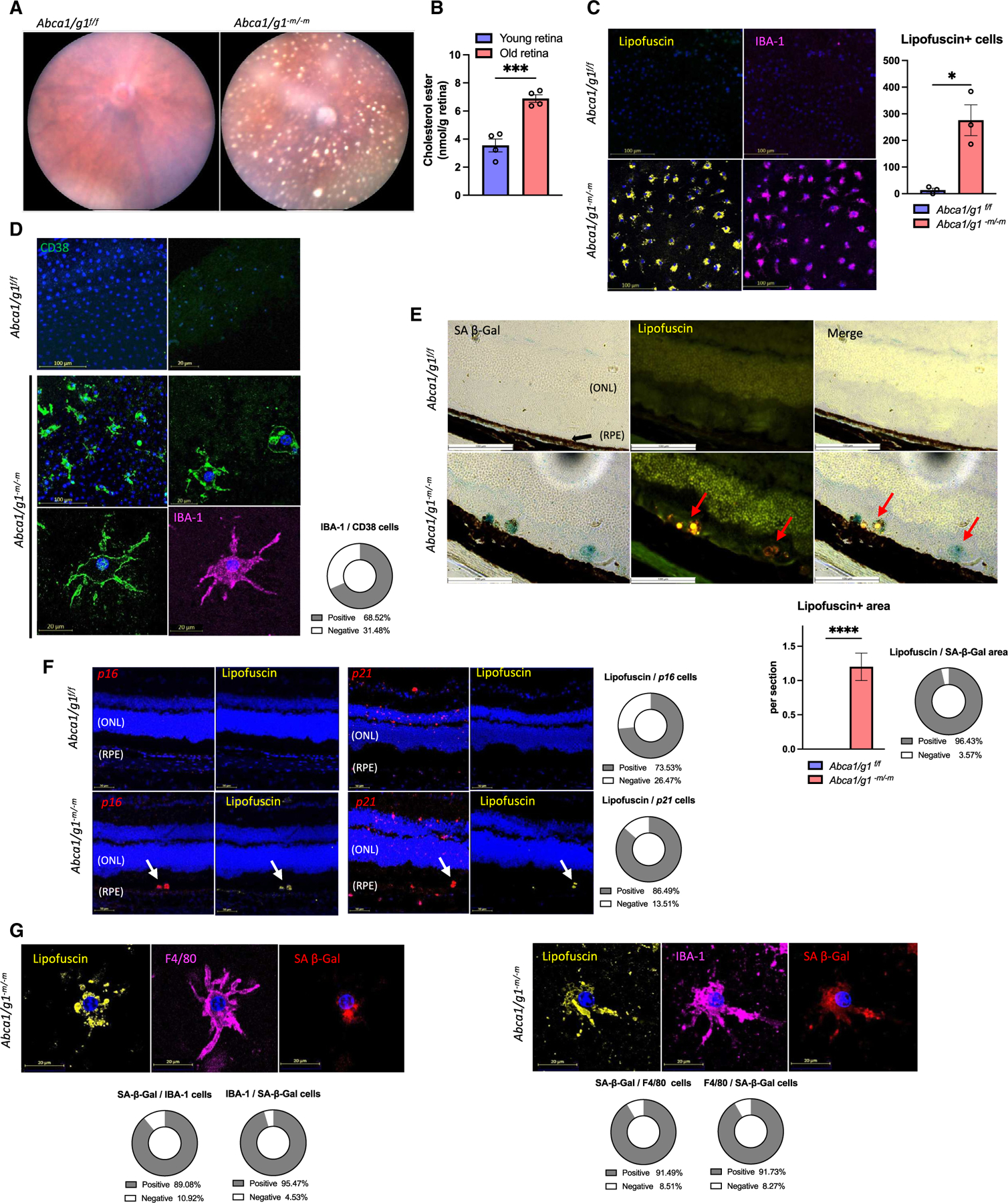
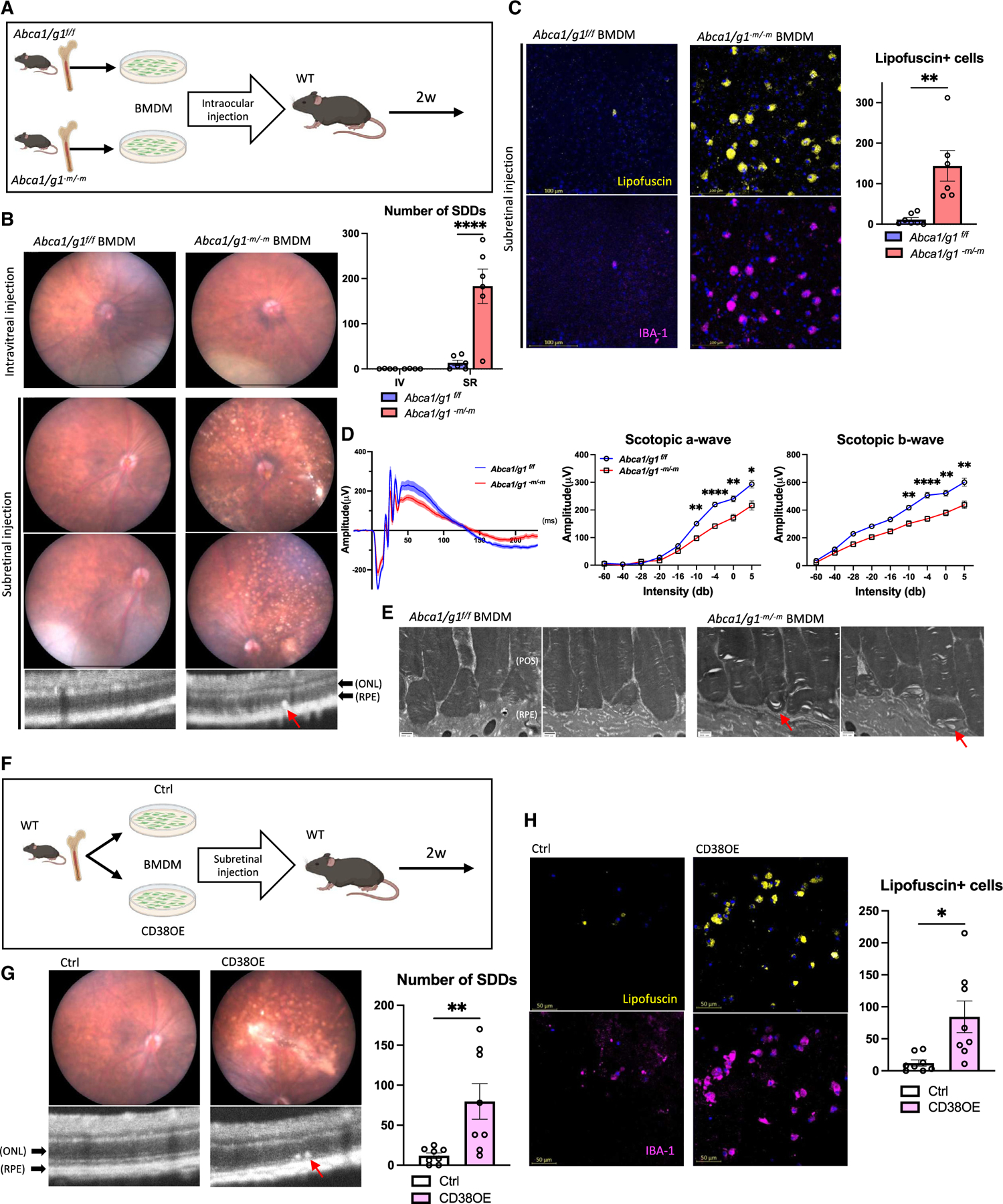
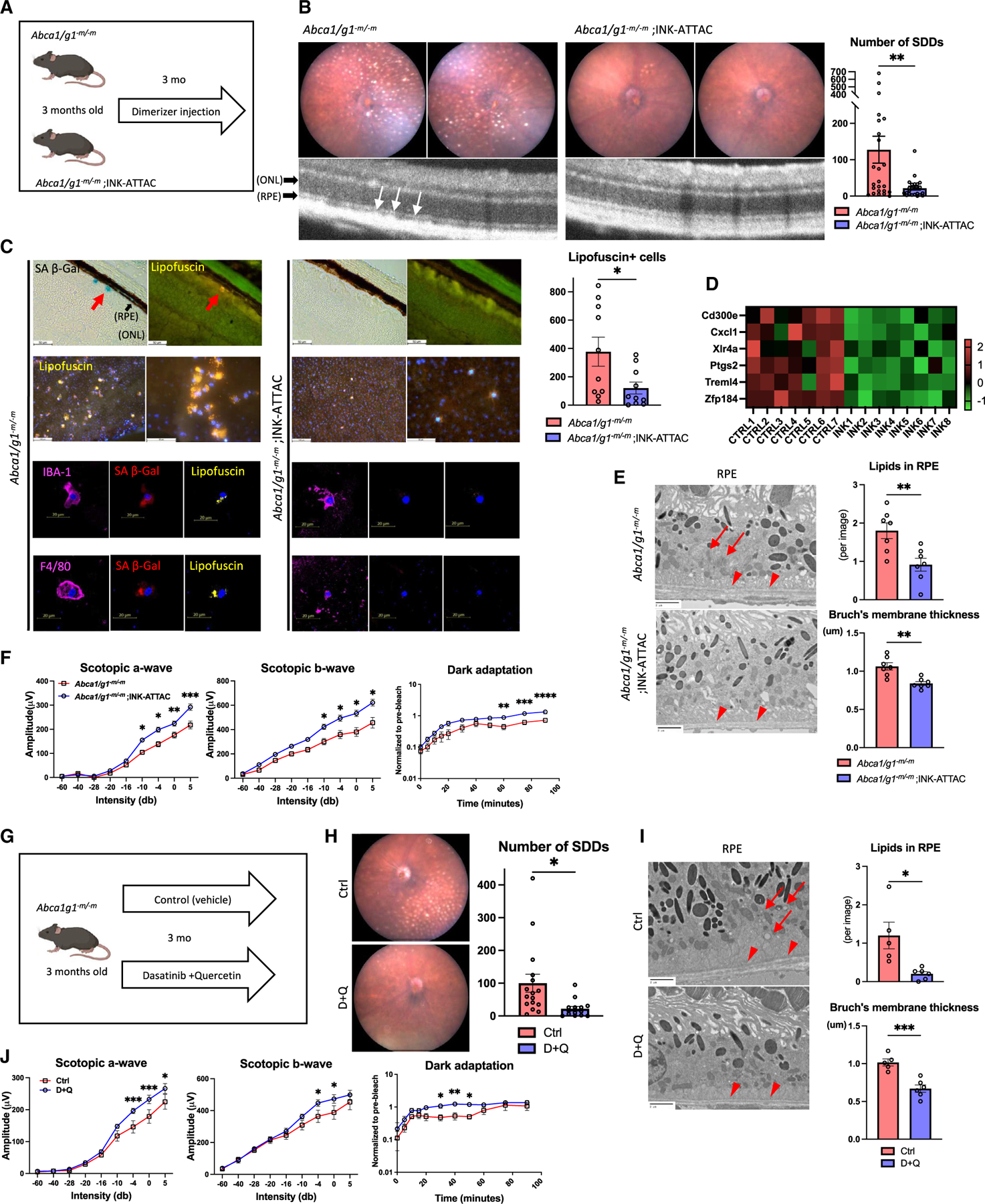
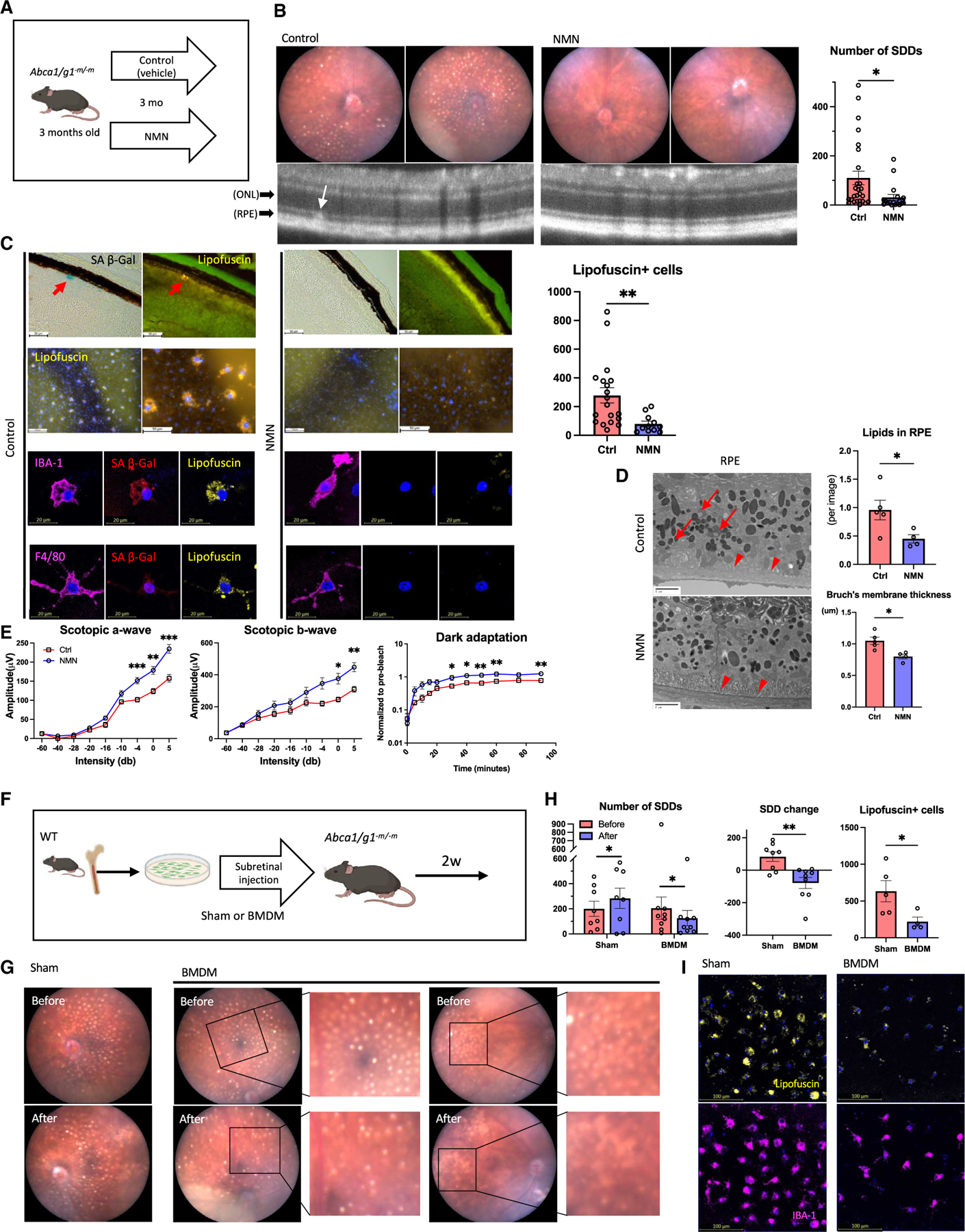
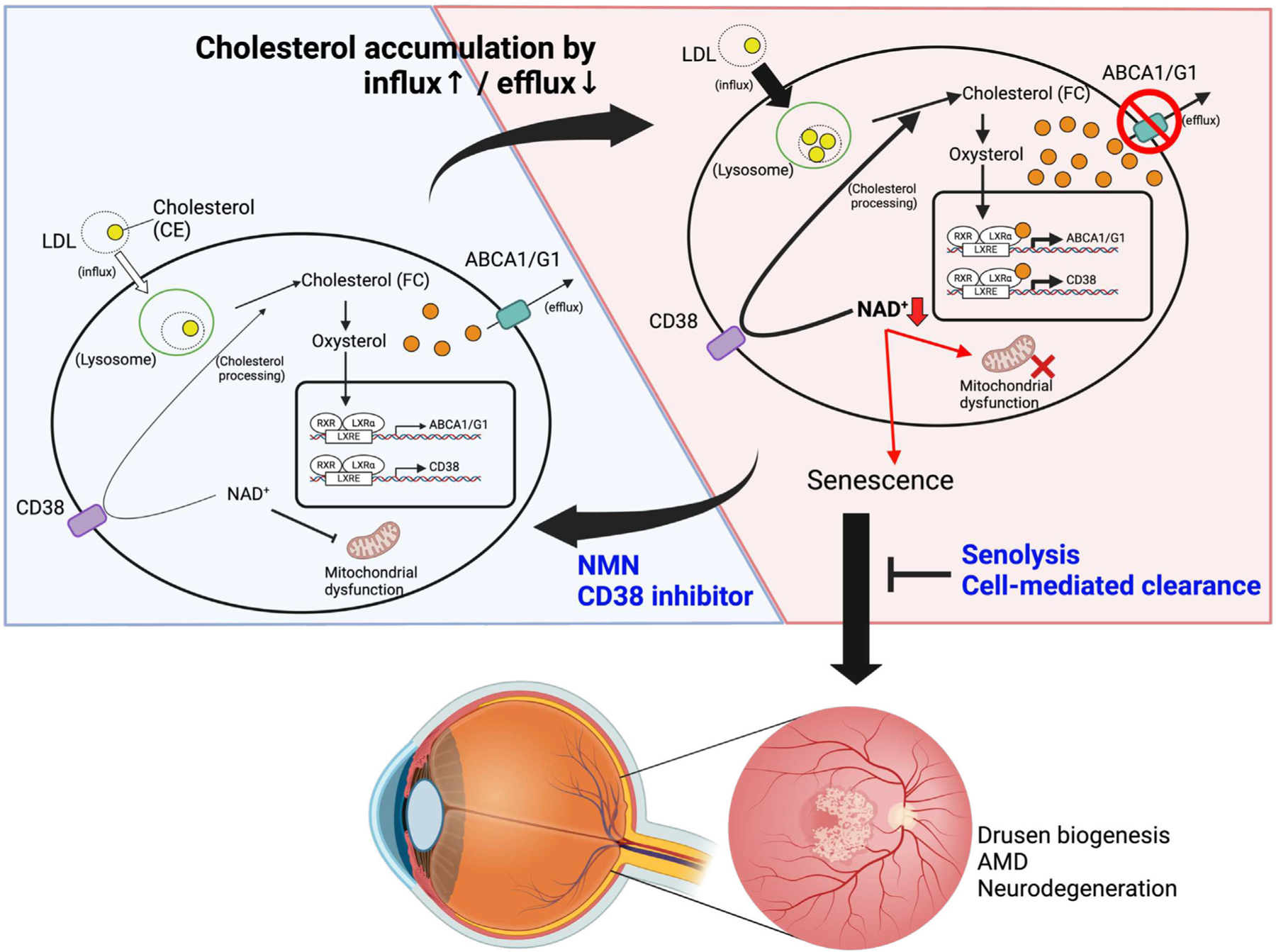
Although dysregulated cholesterol metabolism predisposes aging tissues to inflammation and a plethora of diseases, the underlying molecular mechanism remains poorly defined. Here, we show that metabolic and genotoxic stresses, convergently acting through liver X nuclear receptor, upregulate CD38 to promote lysosomal cholesterol efflux, leading to nicotinamide adenine dinucleotide (NAD+) depletion in macrophages. Cholesterol-mediated NAD+ depletion induces macrophage senescence, promoting key features of age-related macular degeneration (AMD), including subretinal lipid deposition and neurodegeneration. NAD+ augmentation reverses cellular senescence and macrophage dysfunction, preventing the development of AMD phenotype. Genetic and pharmacological senolysis protect against the development of AMD and neurodegeneration. Subretinal administration of healthy macrophages promotes the clearance of senescent macrophages, reversing the AMD disease burden. Thus, NAD+ deficit induced by excess intracellular cholesterol is the converging mechanism of macrophage senescence and a causal process underlying age-related neurodegeneration.
Keywords: CD38; CP: Immunology; CP: Metabolism; NAD(+); NMN; age-related macular degeneration; cellular senescence; cholesterol efflux; neurodegeneration; nicotinamide adenine dinucleotide; nicotinamide mononucleotide.
Copyright © 2024 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests R.S.A. is a co-founder of Metro Midwest Biotech. M.Y. receives patent-licensing fees from Institute for Research on Productive Aging.
Figures
References
-
- Coppé JP, Patil CK, Rodier F, Sun Y, Muñoz DP, Goldstein J, Nelson PS, Desprez PY, and Campisi J (2008). Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol. 6, 2853–2868. 10.1371/journal.pbio.0060301. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
