

The genetic landscape of a metabolic interaction
- PMID: 38637543
- PMCID: PMC11026382
- DOI: 10.1038/s41467-024-47671-0
The genetic landscape of a metabolic interaction
Abstract
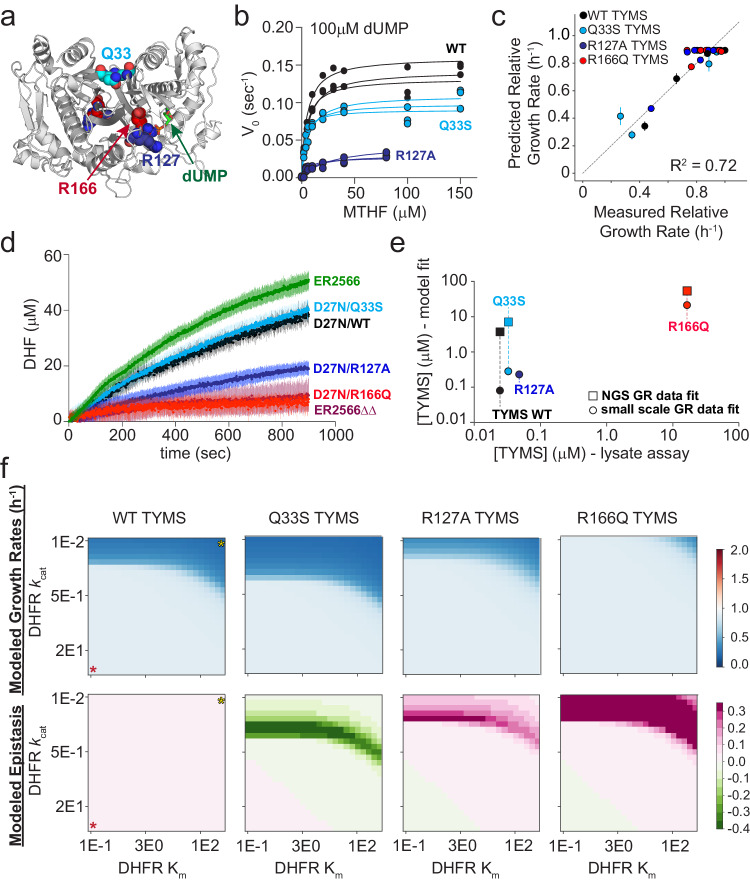
While much prior work has explored the constraints on protein sequence and evolution induced by physical protein-protein interactions, the sequence-level constraints emerging from non-binding functional interactions in metabolism remain unclear. To quantify how variation in the activity of one enzyme constrains the biochemical parameters and sequence of another, we focus on dihydrofolate reductase (DHFR) and thymidylate synthase (TYMS), a pair of enzymes catalyzing consecutive reactions in folate metabolism. We use deep mutational scanning to quantify the growth rate effect of 2696 DHFR single mutations in 3 TYMS backgrounds under conditions selected to emphasize biochemical epistasis. Our data are well-described by a relatively simple enzyme velocity to growth rate model that quantifies how metabolic context tunes enzyme mutational tolerance. Together our results reveal the structural distribution of epistasis in a metabolic enzyme and establish a foundation for the design of multi-enzyme systems.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures






Update of
-
The Genetic Landscape of a Metabolic Interaction.bioRxiv [Preprint]. 2023 Aug 17:2023.05.28.542639. doi: 10.1101/2023.05.28.542639. bioRxiv. 2023. Update in: Nat Commun. 2024 Apr 18;15(1):3351. doi: 10.1038/s41467-024-47671-0. PMID: 37645784 Free PMC article. Updated. Preprint.
References
-
- Kemble, H. E. et al. Flux, toxicity and protein expression costs shape genetic interaction in a metabolic pathway. http://biorxiv.org/lookup/doi/10.1101/362327 (2018) 10.1101/362327. - DOI
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
