

Genetic modifiers of rare variants in monogenic developmental disorder loci
- PMID: 38637616
- PMCID: PMC11096126
- DOI: 10.1038/s41588-024-01710-0
Genetic modifiers of rare variants in monogenic developmental disorder loci
Abstract
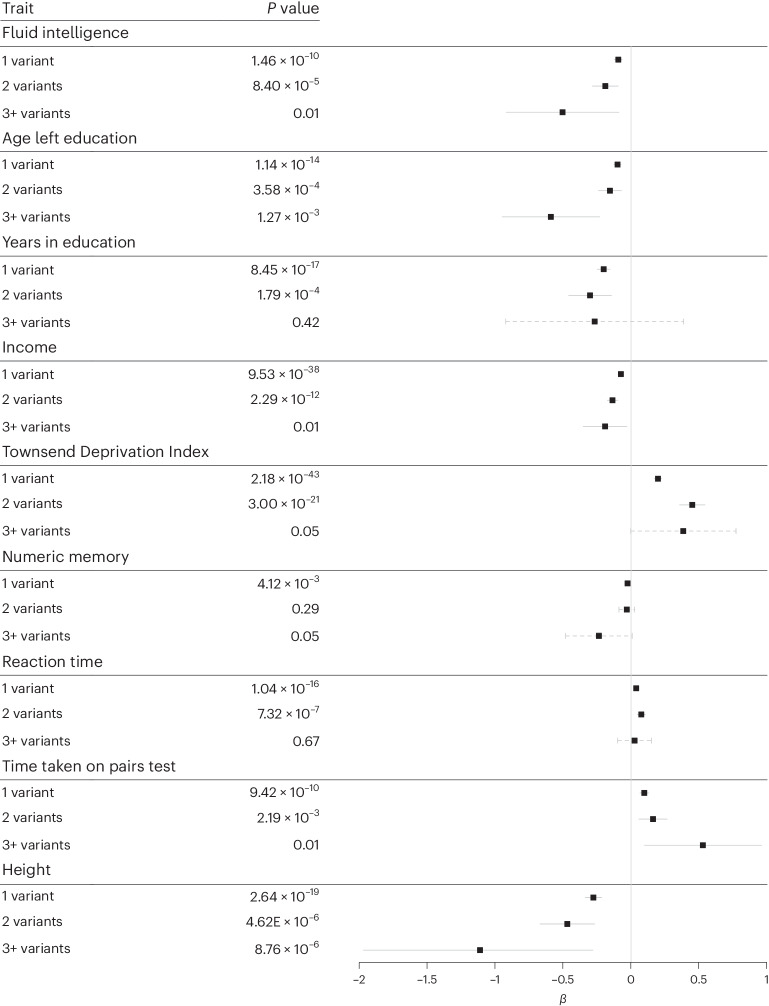
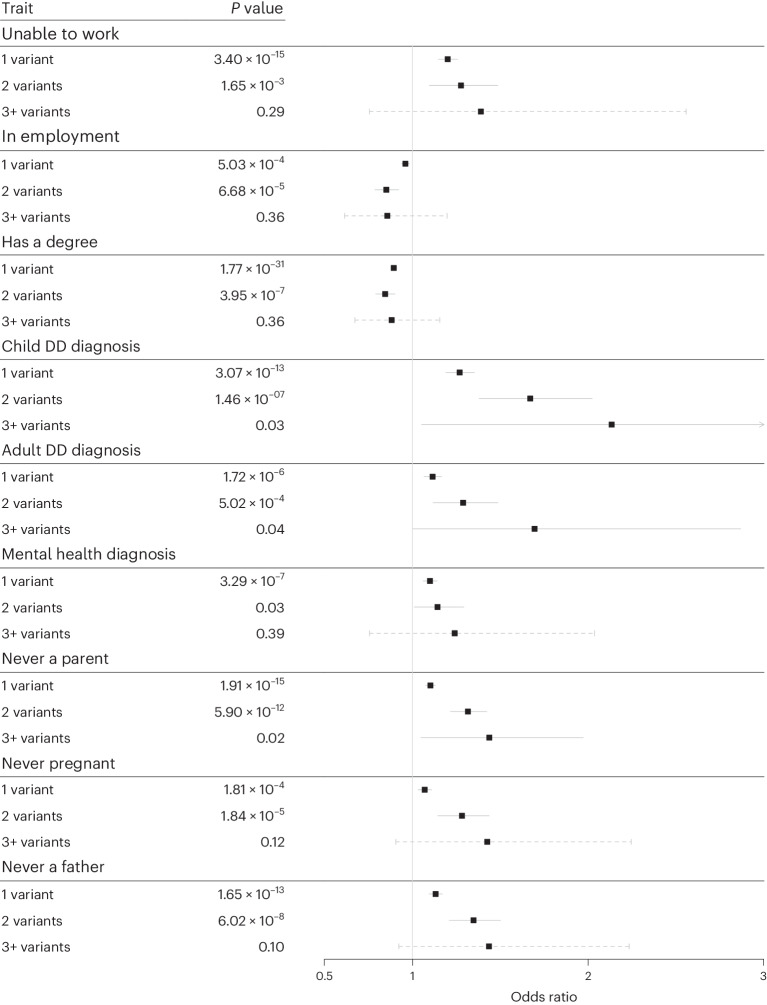
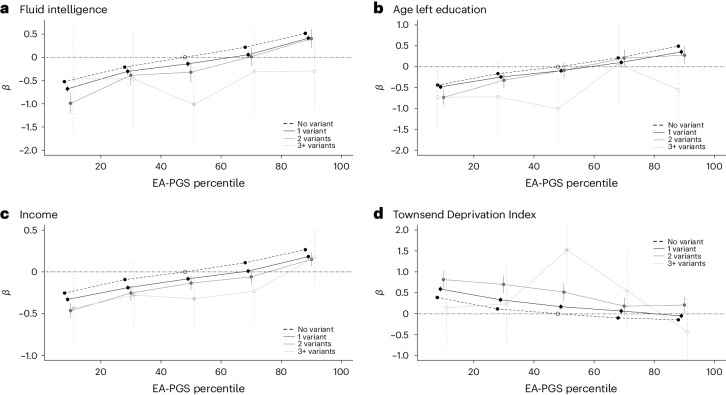
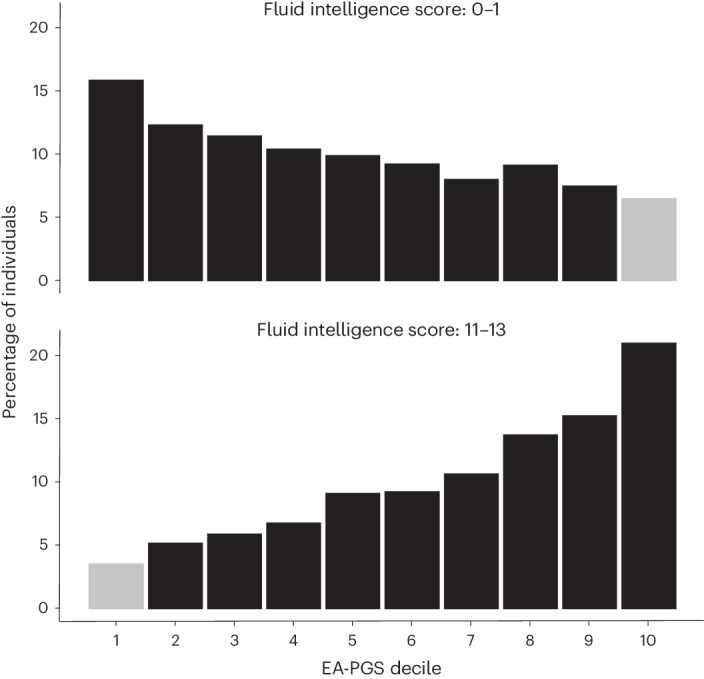
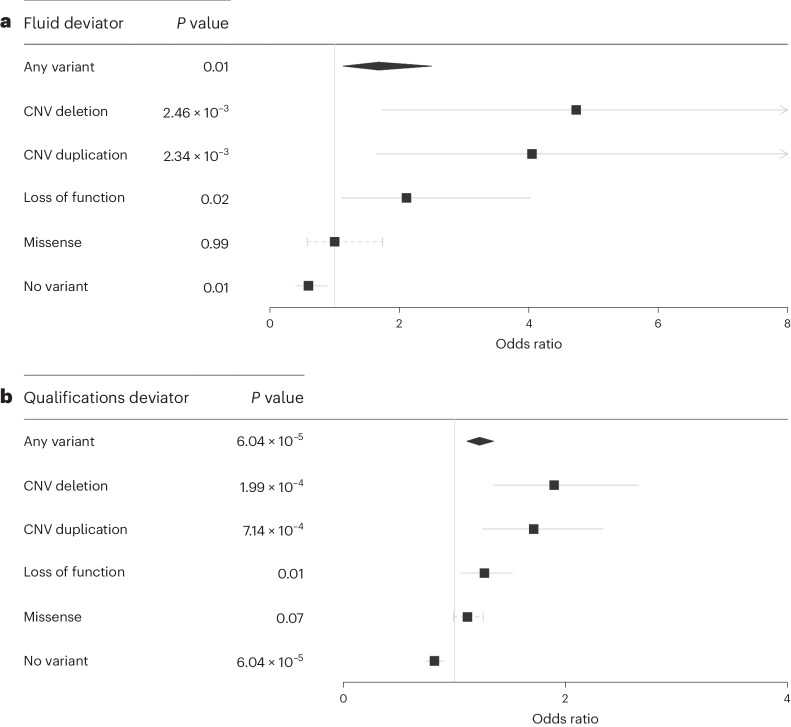
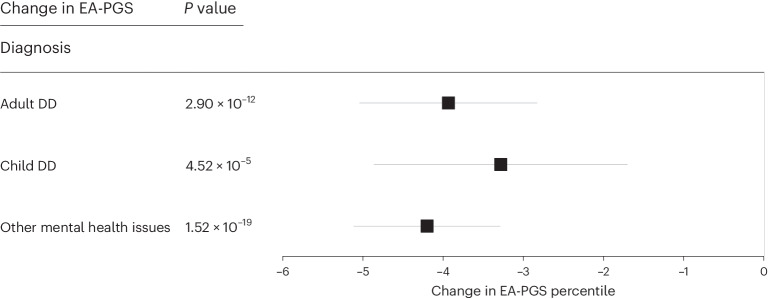
Rare damaging variants in a large number of genes are known to cause monogenic developmental disorders (DDs) and have also been shown to cause milder subclinical phenotypes in population cohorts. Here, we show that carrying multiple (2-5) rare damaging variants across 599 dominant DD genes has an additive adverse effect on numerous cognitive and socioeconomic traits in UK Biobank, which can be partially counterbalanced by a higher educational attainment polygenic score (EA-PGS). Phenotypic deviators from expected EA-PGS could be partly explained by the enrichment or depletion of rare DD variants. Among carriers of rare DD variants, those with a DD-related clinical diagnosis had a substantially lower EA-PGS and more severe phenotype than those without a clinical diagnosis. Our results suggest that the overall burden of both rare and common variants can modify the expressivity of a phenotype, which may then influence whether an individual reaches the threshold for clinical disease.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
