

GENESIS CGDYN: large-scale coarse-grained MD simulation with dynamic load balancing for heterogeneous biomolecular systems
- PMID: 38643169
- PMCID: PMC11032353
- DOI: 10.1038/s41467-024-47654-1
GENESIS CGDYN: large-scale coarse-grained MD simulation with dynamic load balancing for heterogeneous biomolecular systems
Abstract
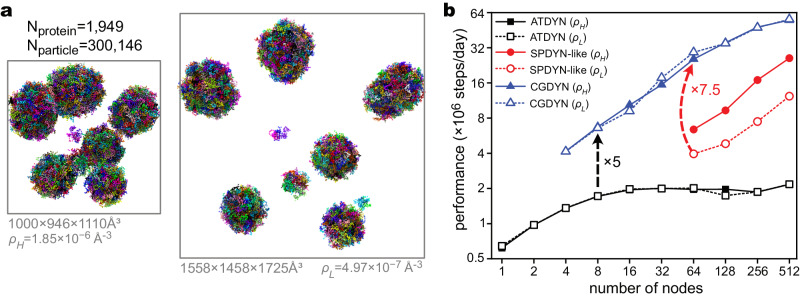
Residue-level coarse-grained (CG) molecular dynamics (MD) simulation is widely used to investigate slow biological processes that involve multiple proteins, nucleic acids, and their complexes. Biomolecules in a large simulation system are distributed non-uniformly, limiting computational efficiency with conventional methods. Here, we develop a hierarchical domain decomposition scheme with dynamic load balancing for heterogeneous biomolecular systems to keep computational efficiency even after drastic changes in particle distribution. These schemes are applied to the dynamics of intrinsically disordered protein (IDP) droplets. During the fusion of two droplets, we find that the changes in droplet shape correlate with the mixing of IDP chains. Additionally, we simulate large systems with multiple IDP droplets, achieving simulation sizes comparable to those observed in microscopy. In our MD simulations, we directly observe Ostwald ripening, a phenomenon where small droplets dissolve and their molecules redeposit into larger droplets. These methods have been implemented in CGDYN of the GENESIS software, offering a tool for investigating mesoscopic biological processes using the residue-level CG models.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interest.
Figures



Similar articles
-
Implementation of residue-level coarse-grained models in GENESIS for large-scale molecular dynamics simulations.PLoS Comput Biol. 2022 Apr 5;18(4):e1009578. doi: 10.1371/journal.pcbi.1009578. eCollection 2022 Apr. PLoS Comput Biol. 2022. PMID: 35381009 Free PMC article.
-
Systematic methods for defining coarse-grained maps in large biomolecules.Adv Exp Med Biol. 2015;827:33-48. doi: 10.1007/978-94-017-9245-5_4. Adv Exp Med Biol. 2015. PMID: 25387958
-
RedMDStream: Parameterization and Simulation Toolbox for Coarse-Grained Molecular Dynamics Models.Biophys J. 2015 Apr 21;108(8):1843-7. doi: 10.1016/j.bpj.2015.03.023. Biophys J. 2015. PMID: 25902423 Free PMC article.
-
Coarse-grained molecular simulations of large biomolecules.Curr Opin Struct Biol. 2012 Apr;22(2):130-7. doi: 10.1016/j.sbi.2012.01.010. Epub 2012 Feb 23. Curr Opin Struct Biol. 2012. PMID: 22365574 Review.
-
Recent Advances in Coarse-Grained Models for Biomolecules and Their Applications.Int J Mol Sci. 2019 Aug 1;20(15):3774. doi: 10.3390/ijms20153774. Int J Mol Sci. 2019. PMID: 31375023 Free PMC article. Review.
Cited by
-
A coarse-grained model for disordered proteins under crowded conditions.Protein Sci. 2025 Aug;34(8):e70232. doi: 10.1002/pro.70232. Protein Sci. 2025. PMID: 40671581 Free PMC article.
-
Recent Progress in Modeling and Simulation of Biomolecular Crowding and Condensation Inside Cells.J Chem Inf Model. 2024 Dec 23;64(24):9063-9081. doi: 10.1021/acs.jcim.4c01520. Epub 2024 Dec 11. J Chem Inf Model. 2024. PMID: 39660892 Free PMC article. Review.
-
Toward understanding biomolecular materials comprising intrinsically disordered proteins via simulation and experiment.Mol Syst Des Eng. 2025 Apr 25;10(7):502-518. doi: 10.1039/d4me00197d. eCollection 2025 Jun 30. Mol Syst Des Eng. 2025. PMID: 40386766 Free PMC article. Review.
-
Parallelization of particle-based reaction-diffusion simulations using MPI.bioRxiv [Preprint]. 2024 Dec 10:2024.12.06.627287. doi: 10.1101/2024.12.06.627287. bioRxiv. 2024. Update in: J Comput Chem. 2025 May 30;46(14):e70132. doi: 10.1002/jcc.70132. PMID: 39713431 Free PMC article. Updated. Preprint.
-
GENESIS 2.1: High-Performance Molecular Dynamics Software for Enhanced Sampling and Free-Energy Calculations for Atomistic, Coarse-Grained, and Quantum Mechanics/Molecular Mechanics Models.J Phys Chem B. 2024 Jun 27;128(25):6028-6048. doi: 10.1021/acs.jpcb.4c02096. Epub 2024 Jun 14. J Phys Chem B. 2024. PMID: 38876465 Free PMC article.
References
-
- Clementi C, Nymeyer H, Onuchic JN. Topological and energetic factors: what determines the structural details of the transition state ensemble and “en-route” intermediates for protein folding? An investigation for small globular proteins. J. Mol. Biol. 2000;298:937–953. doi: 10.1006/jmbi.2000.3693. - DOI - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
