

C/EBPβ: A transcription factor associated with the irreversible progression of Alzheimer's disease
- PMID: 38644578
- PMCID: PMC11033503
- DOI: 10.1111/cns.14721
C/EBPβ: A transcription factor associated with the irreversible progression of Alzheimer's disease
Abstract
Background: Alzheimer's disease (AD) is a neurodegenerative disorder distinguished by a swift cognitive deterioration accompanied by distinctive pathological hallmarks such as extracellular Aβ (β-amyloid) peptides, neuronal neurofibrillary tangles (NFTs), sustained neuroinflammation, and synaptic degeneration. The elevated frequency of AD cases and its proclivity to manifest at a younger age present a pressing challenge in the quest for novel therapeutic interventions. Numerous investigations have substantiated the involvement of C/EBPβ in the progression of AD pathology, thus indicating its potential as a therapeutic target for AD treatment.
Aims: Several studies have demonstrated an elevation in the expression level of C/EBPβ among individuals afflicted with AD. Consequently, this review predominantly delves into the association between C/EBPβ expression and the pathological progression of Alzheimer's disease, elucidating its underlying molecular mechanism, and pointing out the possibility that C/EBPβ can be a new therapeutic target for AD.
Methods: A systematic literature search was performed across multiple databases, including PubMed, Google Scholar, and so on, utilizing predetermined keywords and MeSH terms, without temporal constraints. The inclusion criteria encompassed diverse study designs, such as experimental, case-control, and cohort studies, restricted to publications in the English language, while conference abstracts and unpublished sources were excluded.
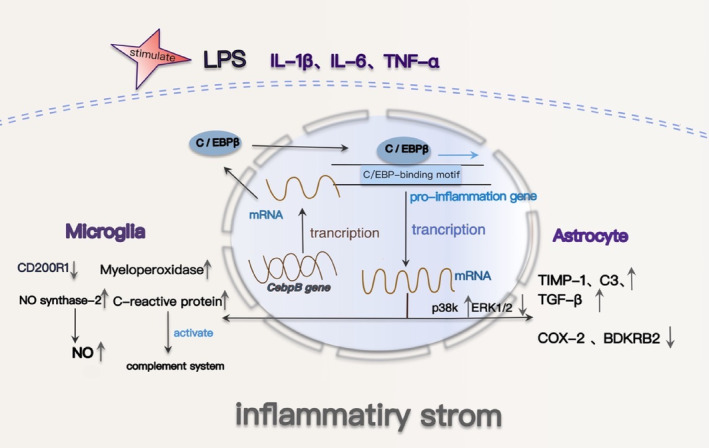
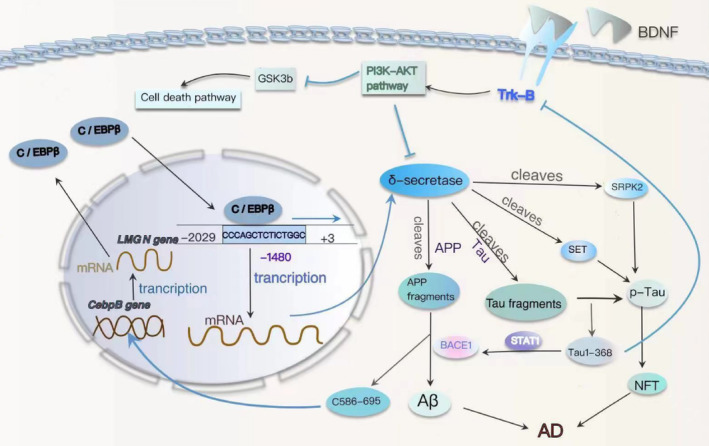
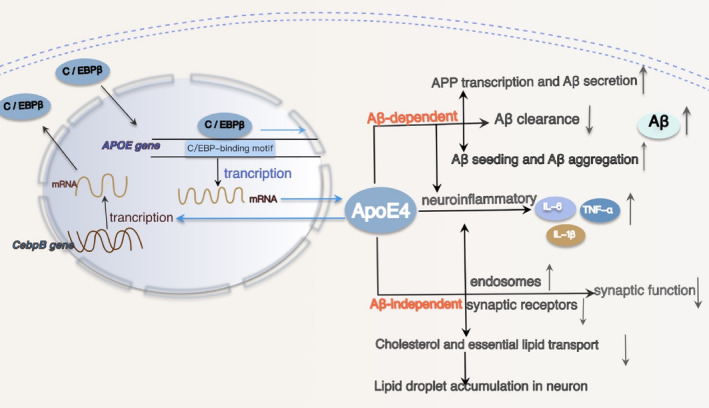
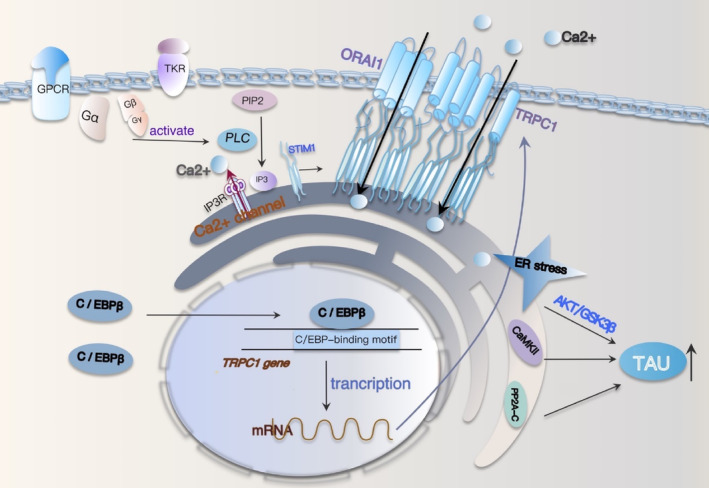
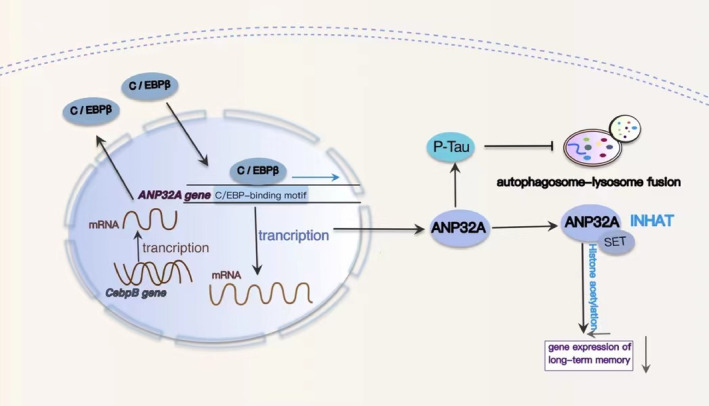
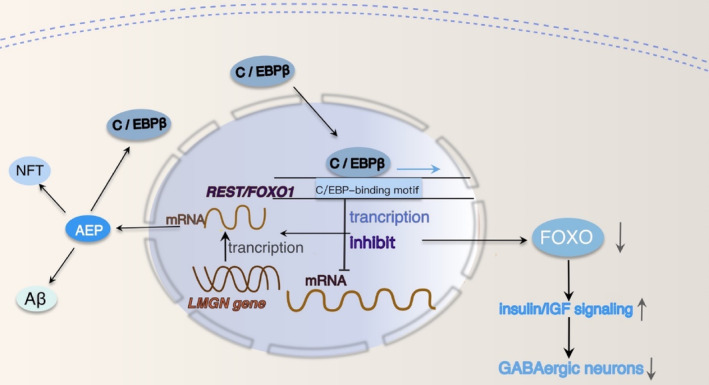
Results: Overexpression of C/EBPβ exacerbates the pathological features of AD, primarily by promoting neuroinflammation and mediating the transcriptional regulation of key molecular pathways, including δ-secretase, apolipoprotein E4 (APOE4), acidic leucine-rich nuclear phosphoprotein-32A (ANP32A), transient receptor potential channel 1 (TRPC1), and Forkhead BoxO (FOXO).
Discussion: The correlation between overexpression of C/EBPβ and the pathological development of AD, along with its molecular mechanisms, is evident. Investigating the pathways through which C/EBPβ regulates the development of AD reveals numerous multiple vicious cycle pathways exacerbating the pathological progression of the disease. Furthermore, the exacerbation of pathological progression due to C/EBPβ overexpression and its molecular mechanism is not limited to AD but also extends to other neurodegenerative diseases such as amyotrophic lateral sclerosis (ALS), Parkinson's disease (PD), and multiple sclerosis (MS).
Conclusion: The overexpression of C/EBPβ accelerates the irreversible progression of AD pathophysiology. Additionally, C/EBPβ plays a crucial role in mediating multiple pathways linked to AD pathology, some of which engender vicious cycles, leading to the establishment of feedback mechanisms. To sum up, targeting C/EBPβ could hold promise as a therapeutic strategy not only for AD but also for other degenerative diseases.
Keywords: Alzheimer's disease; C/EBPβ; neurodegenerative disease; therapy; transcription.
© 2024 The Authors. CNS Neuroscience & Therapeutics published by John Wiley & Sons Ltd.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Co-Aggregation of Syndecan-3 with β-Amyloid Aggravates Neuroinflammation and Cognitive Impairment in 5×FAD Mice.Int J Mol Sci. 2025 Jun 8;26(12):5502. doi: 10.3390/ijms26125502. Int J Mol Sci. 2025. PMID: 40564963 Free PMC article.
-
Short-Term Memory Impairment.2024 Jun 8. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. 2024 Jun 8. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan–. PMID: 31424720 Free Books & Documents.
-
The Black Book of Psychotropic Dosing and Monitoring.Psychopharmacol Bull. 2024 Jul 8;54(3):8-59. Psychopharmacol Bull. 2024. PMID: 38993656 Free PMC article. Review.
-
CSF tau and the CSF tau/ABeta ratio for the diagnosis of Alzheimer's disease dementia and other dementias in people with mild cognitive impairment (MCI).Cochrane Database Syst Rev. 2017 Mar 22;3(3):CD010803. doi: 10.1002/14651858.CD010803.pub2. Cochrane Database Syst Rev. 2017. PMID: 28328043 Free PMC article.
-
Selegiline for Alzheimer's disease.Cochrane Database Syst Rev. 2003;(1):CD000442. doi: 10.1002/14651858.CD000442. Cochrane Database Syst Rev. 2003. PMID: 12535396
Cited by
-
Programmed cell death-related prognostic genes mediate dysregulation of the immune microenvironment in triple-negative breast cancer.Front Immunol. 2025 Mar 12;16:1563630. doi: 10.3389/fimmu.2025.1563630. eCollection 2025. Front Immunol. 2025. PMID: 40145099 Free PMC article.
-
Buyang Huanwu Decoction Modulates the Gut Microbiota-C/EBPβ/AEP Axis to Ameliorate Cognitive Impairment in Alzheimer's Disease Mice.CNS Neurosci Ther. 2025 Jun;31(6):e70480. doi: 10.1111/cns.70480. CNS Neurosci Ther. 2025. PMID: 40546239 Free PMC article.
-
Intermittent Hypoxia Induces Cognitive Dysfunction and Hippocampal Gene Expression Changes in a Mouse Model of Obstructive Sleep Apnea.Int J Mol Sci. 2025 Aug 3;26(15):7495. doi: 10.3390/ijms26157495. Int J Mol Sci. 2025. PMID: 40806623 Free PMC article.
-
Microglial C/EBPβ-Fcgr1 regulatory axis blocking inhibits microglial pyroptosis and improves neurological recovery.J Neuroinflammation. 2025 Jan 31;22(1):29. doi: 10.1186/s12974-025-03362-1. J Neuroinflammation. 2025. PMID: 39891259 Free PMC article.
-
GSK3: A potential target and pending issues for treatment of Alzheimer's disease.CNS Neurosci Ther. 2024 Jul;30(7):e14818. doi: 10.1111/cns.14818. CNS Neurosci Ther. 2024. PMID: 38946682 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
