

Molecular diversity in isocitrate dehydrogenase-wild-type glioblastoma
- PMID: 38646145
- PMCID: PMC11032202
- DOI: 10.1093/braincomms/fcae108
Molecular diversity in isocitrate dehydrogenase-wild-type glioblastoma
Abstract
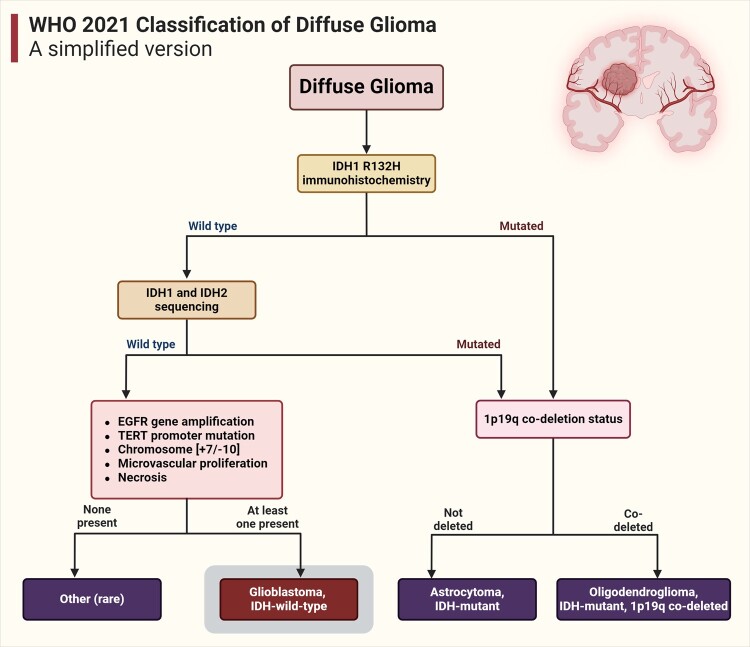
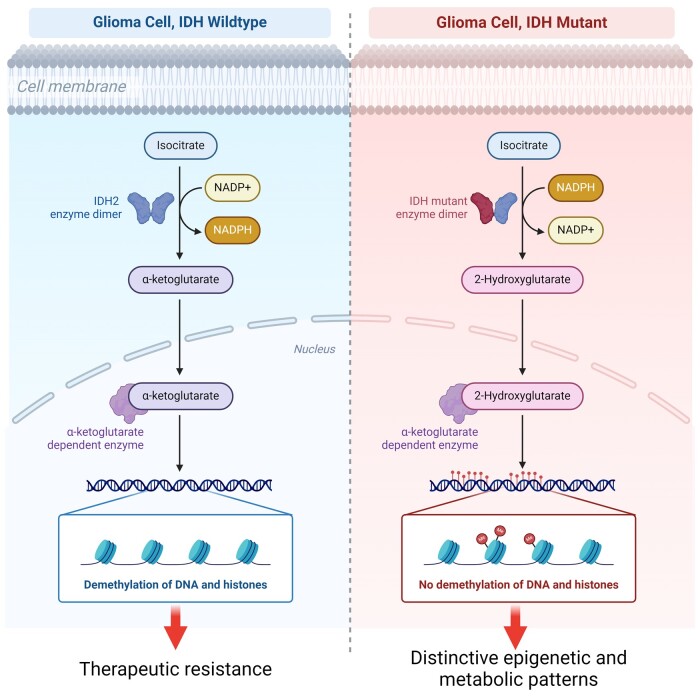
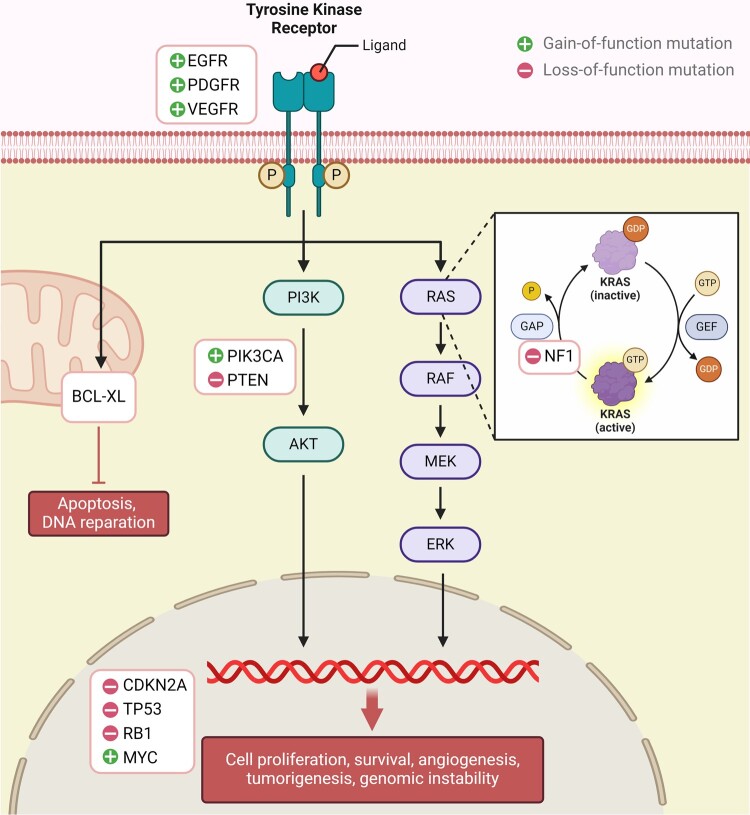
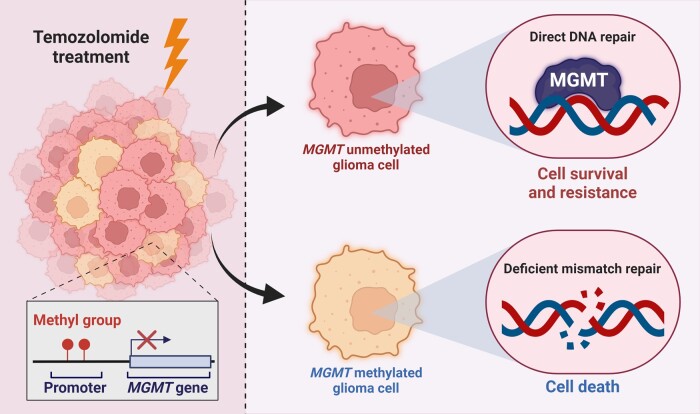
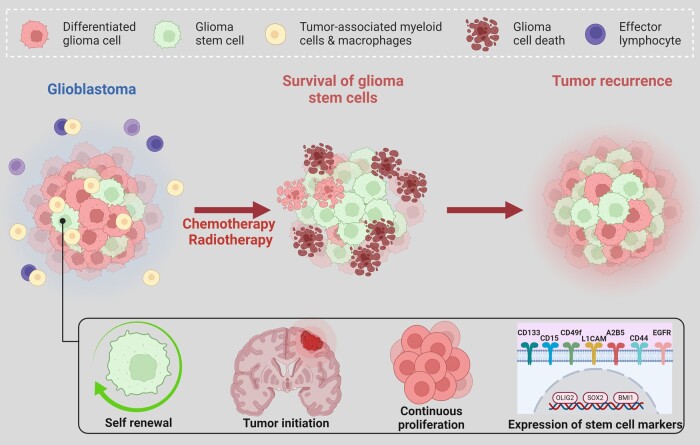
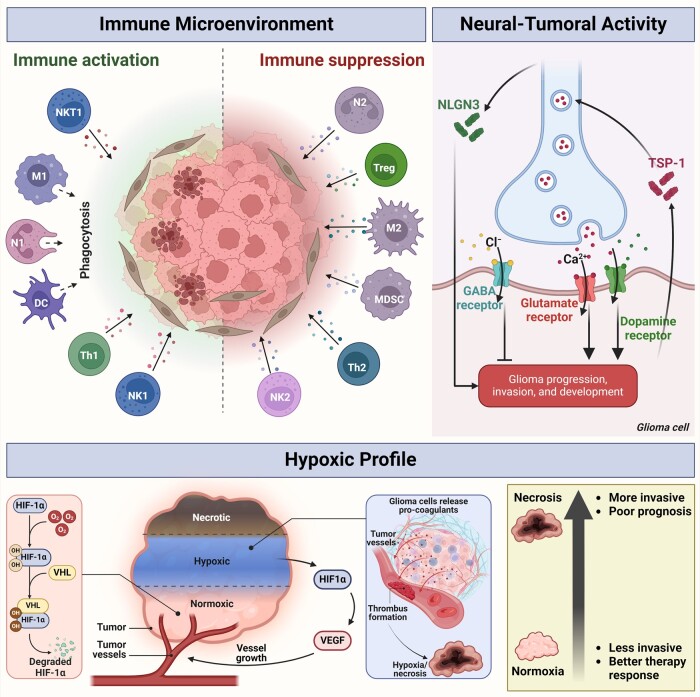
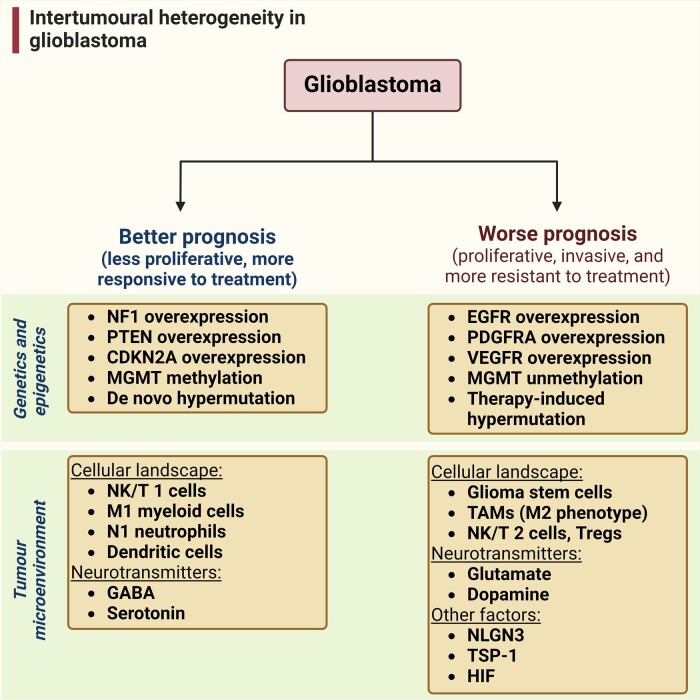
In the dynamic landscape of glioblastoma, the 2021 World Health Organization Classification of Central Nervous System tumours endeavoured to establish biological homogeneity, yet isocitrate dehydrogenase-wild-type (IDH-wt) glioblastoma persists as a tapestry of clinical and molecular diversity. Intertumoural heterogeneity in IDH-wt glioblastoma presents a formidable challenge in treatment strategies. Recent strides in genetics and molecular biology have enhanced diagnostic precision, revealing distinct subtypes and invasive patterns that influence survival in patients with IDH-wt glioblastoma. Genetic and molecular biomarkers, such as the overexpression of neurofibromin 1, phosphatase and tensin homolog and/or cyclin-dependent kinase inhibitor 2A, along with specific immune cell abundance and neurotransmitters, correlate with favourable outcomes. Conversely, increased expression of epidermal growth factor receptor tyrosine kinase, platelet-derived growth factor receptor alpha and/or vascular endothelial growth factor receptor, coupled with the prevalence of glioma stem cells, tumour-associated myeloid cells, regulatory T cells and exhausted effector cells, signifies an unfavourable prognosis. The methylation status of O6-methylguanine-DNA methyltransferase and the influence of microenvironmental factors and neurotransmitters further shape treatment responses. Understanding intertumoural heterogeneity is complemented by insights into intratumoural dynamics and cellular interactions within the tumour microenvironment. Glioma stem cells and immune cell composition significantly impact progression and outcomes, emphasizing the need for personalized therapies targeting pro-tumoural signalling pathways and resistance mechanisms. A successful glioblastoma management demands biomarker identification, combination therapies and a nuanced approach considering intratumoural variability. These advancements herald a transformative era in glioblastoma comprehension and treatment.
Keywords: IDH-wild-type; glioblastoma; heterogeneity; machine learning; neuroimaging.
© The Author(s) 2024. Published by Oxford University Press on behalf of the Guarantors of Brain.
Conflict of interest statement
The authors report no competing interests.
Figures

Similar articles
-
Use of telomerase promoter mutations to mark specific molecular subsets with reciprocal clinical behavior in IDH mutant and IDH wild-type diffuse gliomas.J Neurosurg. 2018 Apr;128(4):1102-1114. doi: 10.3171/2016.11.JNS16973. Epub 2017 Jun 16. J Neurosurg. 2018. PMID: 28621624
-
Association of Maximal Extent of Resection of Contrast-Enhanced and Non-Contrast-Enhanced Tumor With Survival Within Molecular Subgroups of Patients With Newly Diagnosed Glioblastoma.JAMA Oncol. 2020 Apr 1;6(4):495-503. doi: 10.1001/jamaoncol.2019.6143. JAMA Oncol. 2020. PMID: 32027343 Free PMC article.
-
Telomerase reverse transcriptase promoter mutation- and O6-methylguanine DNA methyltransferase promoter methylation-mediated sensitivity to temozolomide in isocitrate dehydrogenase-wild-type glioblastoma: is there a link?Eur J Cancer. 2021 Apr;147:84-94. doi: 10.1016/j.ejca.2021.01.014. Epub 2021 Feb 22. Eur J Cancer. 2021. PMID: 33631540
-
Biomarkers for glioblastoma multiforme: status quo.J Clin Transl Res. 2016 Mar 27;2(1):3-10. eCollection 2016 Apr 15. J Clin Transl Res. 2016. PMID: 30873456 Free PMC article. Review.
-
New hints towards a precision medicine strategy for IDH wild-type glioblastoma.Ann Oncol. 2020 Dec;31(12):1679-1692. doi: 10.1016/j.annonc.2020.08.2336. Epub 2020 Sep 9. Ann Oncol. 2020. PMID: 32918998 Review.
Cited by
-
RANO 2.0: critical updates and practical considerations for radiological assessment in neuro-oncology.Jpn J Radiol. 2025 Jun 30. doi: 10.1007/s11604-025-01821-6. Online ahead of print. Jpn J Radiol. 2025. PMID: 40586994 Review.
-
Therapeutic Targets in Glioblastoma: Molecular Pathways, Emerging Strategies, and Future Directions.Cells. 2025 Mar 26;14(7):494. doi: 10.3390/cells14070494. Cells. 2025. PMID: 40214448 Free PMC article. Review.
-
Spatial invasion patterns of temporal lobe glioblastoma after complete resection of contrast-enhancing tumor.J Neurooncol. 2025 Jun;173(2):353-360. doi: 10.1007/s11060-025-04991-5. Epub 2025 Mar 5. J Neurooncol. 2025. PMID: 40045105 Free PMC article.
-
Immunotherapy in Glioblastoma: An Overview of Current Status.Clin Pharmacol. 2025 Jul 24;17:185-209. doi: 10.2147/CPAA.S497903. eCollection 2025. Clin Pharmacol. 2025. PMID: 40735070 Free PMC article. Review.
-
Decoding Glioblastoma Heterogeneity: Neuroimaging Meets Machine Learning.Neurosurgery. 2025 Jun 1;96(6):1181-1192. doi: 10.1227/neu.0000000000003260. Epub 2024 Nov 21. Neurosurgery. 2025. PMID: 39570018 Review.
References
-
- Stupp R, Mason WP, van den Bent MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352(10):987–996. - PubMed
-
- Wick W, Gorlia T, Bendszus M, et al. Lomustine and bevacizumab in progressive glioblastoma. N Engl J Med. 2017;377(20):1954–1963. - PubMed
-
- Wann A, Tully PA, Barnes EH, et al. Outcomes after second surgery for recurrent glioblastoma: A retrospective case-control study. J Neurooncol. 2018;137(2):409–415. - PubMed
-
- Burrell RA, McGranahan N, Bartek J, Swanton C. The causes and consequences of genetic heterogeneity in cancer evolution. Nature. 2013;501(7467):338–345. - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Research Materials