

Deep mining of the Sequence Read Archive reveals major genetic innovations in coronaviruses and other nidoviruses of aquatic vertebrates
- PMID: 38648214
- PMCID: PMC11065284
- DOI: 10.1371/journal.ppat.1012163
Deep mining of the Sequence Read Archive reveals major genetic innovations in coronaviruses and other nidoviruses of aquatic vertebrates
Abstract
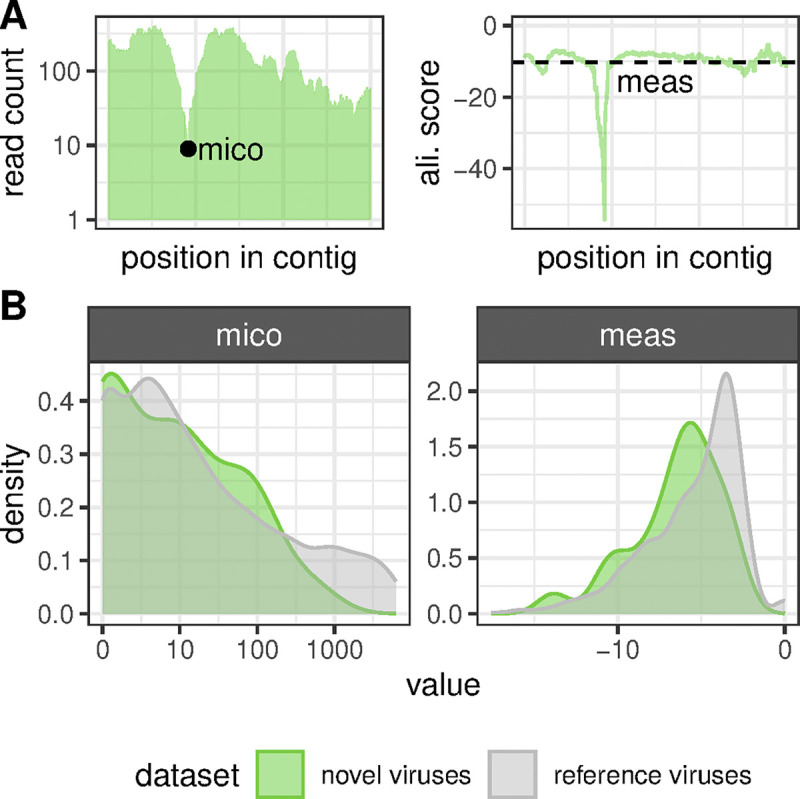
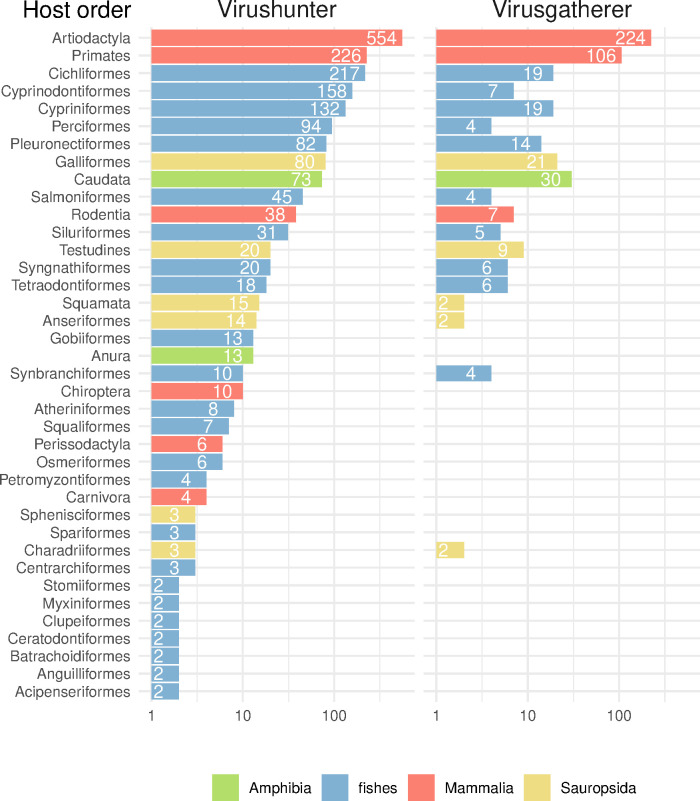
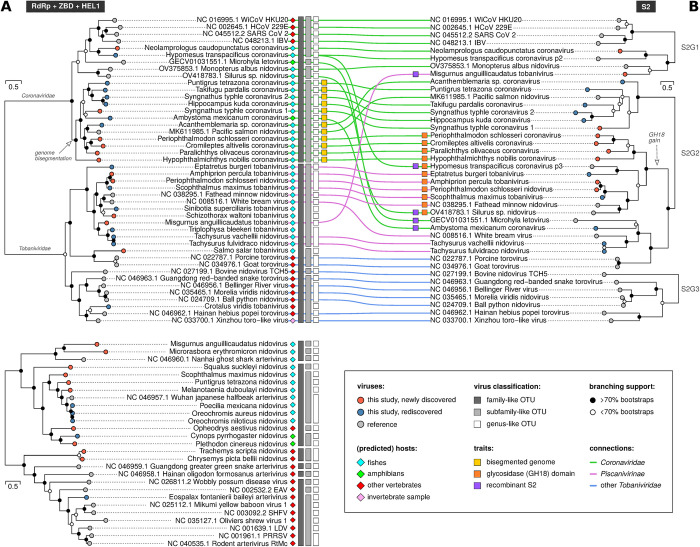
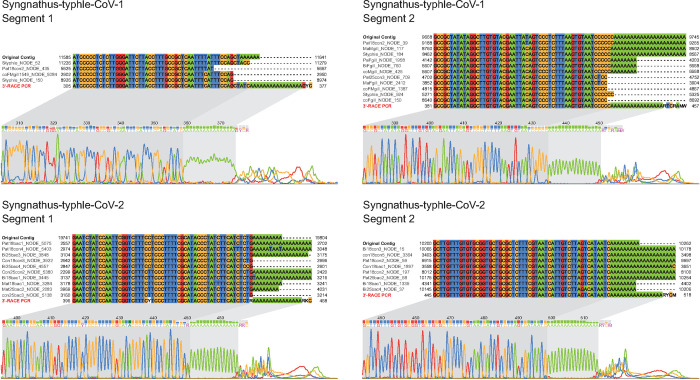
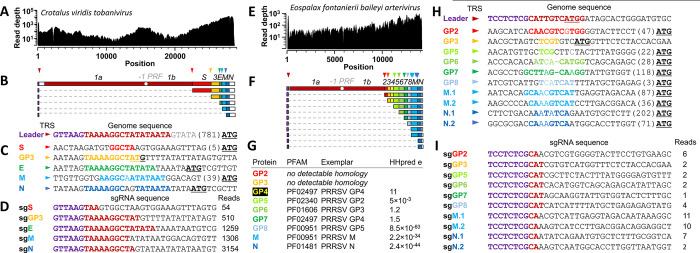
Virus discovery by genomics and metagenomics empowered studies of viromes, facilitated characterization of pathogen epidemiology, and redefined our understanding of the natural genetic diversity of viruses with profound functional and structural implications. Here we employed a data-driven virus discovery approach that directly queries unprocessed sequencing data in a highly parallelized way and involves a targeted viral genome assembly strategy in a wide range of sequence similarity. By screening more than 269,000 datasets of numerous authors from the Sequence Read Archive and using two metrics that quantitatively assess assembly quality, we discovered 40 nidoviruses from six virus families whose members infect vertebrate hosts. They form 13 and 32 putative viral subfamilies and genera, respectively, and include 11 coronaviruses with bisegmented genomes from fishes and amphibians, a giant 36.1 kilobase coronavirus genome with a duplicated spike glycoprotein (S) gene, 11 tobaniviruses and 17 additional corona-, arteri-, cremega-, nanhypo- and nangoshaviruses. Genome segmentation emerged in a single evolutionary event in the monophyletic lineage encompassing the subfamily Pitovirinae. We recovered the bisegmented genome sequences of two coronaviruses from RNA samples of 69 infected fishes and validated the presence of poly(A) tails at both segments using 3'RACE PCR and subsequent Sanger sequencing. We report a genetic linkage between accessory and structural proteins whose phylogenetic relationships and evolutionary distances are incongruent with the phylogeny of replicase proteins. We rationalize these observations in a model of inter-family S recombination involving at least five ancestral corona- and tobaniviruses of aquatic hosts. In support of this model, we describe an individual fish co-infected with members from the families Coronaviridae and Tobaniviridae. Our results expand the scale of the known extraordinary evolutionary plasticity in nidoviral genome architecture and call for revisiting fundamentals of genome expression, virus particle biology, host range and ecology of vertebrate nidoviruses.
Copyright: © 2024 Lauber et al. This is an open access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures

Similar articles
-
Giant RNA genomes: Roles of host, translation elongation, genome architecture, and proteome in nidoviruses.Proc Natl Acad Sci U S A. 2025 Feb 18;122(7):e2413675122. doi: 10.1073/pnas.2413675122. Epub 2025 Feb 10. Proc Natl Acad Sci U S A. 2025. PMID: 39928875 Free PMC article.
-
Characterization of White bream virus reveals a novel genetic cluster of nidoviruses.J Virol. 2006 Dec;80(23):11598-609. doi: 10.1128/JVI.01758-06. Epub 2006 Sep 20. J Virol. 2006. PMID: 16987966 Free PMC article.
-
Discovery of the first insect nidovirus, a missing evolutionary link in the emergence of the largest RNA virus genomes.PLoS Pathog. 2011 Sep;7(9):e1002215. doi: 10.1371/journal.ppat.1002215. Epub 2011 Sep 8. PLoS Pathog. 2011. PMID: 21931546 Free PMC article.
-
Nidovirales: evolving the largest RNA virus genome.Virus Res. 2006 Apr;117(1):17-37. doi: 10.1016/j.virusres.2006.01.017. Epub 2006 Feb 28. Virus Res. 2006. PMID: 16503362 Free PMC article. Review.
-
Properties of Coronavirus and SARS-CoV-2.Malays J Pathol. 2020 Apr;42(1):3-11. Malays J Pathol. 2020. PMID: 32342926 Review.
Cited by
-
Discovery and biological confirmation of a highly divergent Tacaribe virus in metatranscriptomic data from neotropical bats.mSphere. 2024 Oct 29;9(10):e0052024. doi: 10.1128/msphere.00520-24. Epub 2024 Sep 11. mSphere. 2024. PMID: 39258931 Free PMC article.
-
Genome sizes of animal RNA viruses reflect phylogenetic constraints.Virus Evol. 2025 Jan 24;11(1):veaf005. doi: 10.1093/ve/veaf005. eCollection 2025. Virus Evol. 2025. PMID: 39906303 Free PMC article.
-
SegFinder: an automated tool for identifying complete RNA virus genome segments through co-occurrence in multiple sequenced samples.Brief Bioinform. 2025 Jul 2;26(4):bbaf358. doi: 10.1093/bib/bbaf358. Brief Bioinform. 2025. PMID: 40702703 Free PMC article.
-
Insights into the RNA Virome of the Corn Leafhopper Dalbulus maidis, a Major Emergent Threat of Maize in Latin America.Viruses. 2024 Oct 9;16(10):1583. doi: 10.3390/v16101583. Viruses. 2024. PMID: 39459917 Free PMC article.
-
The protein structurome of Orthornavirae and its dark matter.mBio. 2025 Feb 5;16(2):e0320024. doi: 10.1128/mbio.03200-24. Epub 2024 Dec 23. mBio. 2025. PMID: 39714180 Free PMC article.
References
-
- de Groot RJ, Cowley JA, Enjuanes L, Faaberg KS, Perlman S, Rottier PJM, et al.. Order Nidovirales. King AMQ, Adams MJ, Carstens EB, Lefkowitz EJ (editors) Virus Taxonomy, Ninth Report of the International Committee on Taxonomy of Viruses. Amsterdam: Elsevier Academic Press; 2012. pp. 785–795.
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Miscellaneous
