

The Number and Pattern of Viral Genomic Reassortments are not Necessarily Identifiable from Segment Trees
- PMID: 38648521
- PMCID: PMC11152448
- DOI: 10.1093/molbev/msae078
The Number and Pattern of Viral Genomic Reassortments are not Necessarily Identifiable from Segment Trees
Abstract
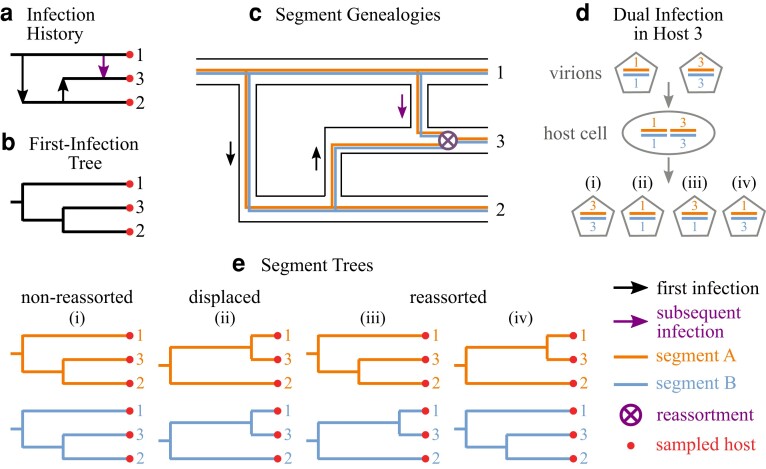
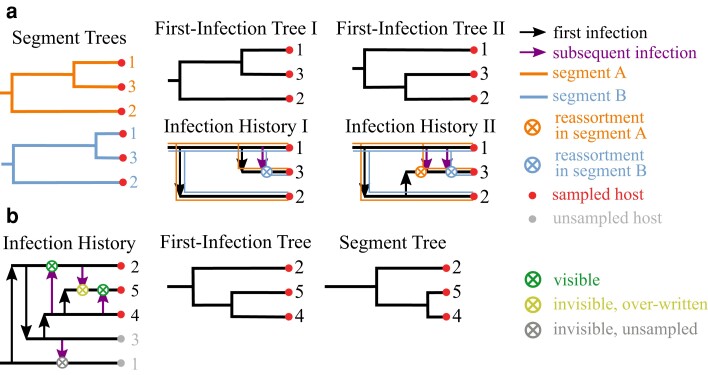
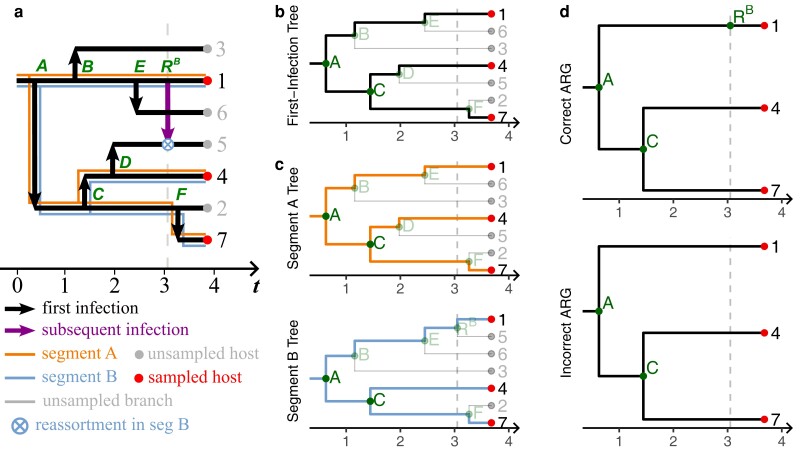
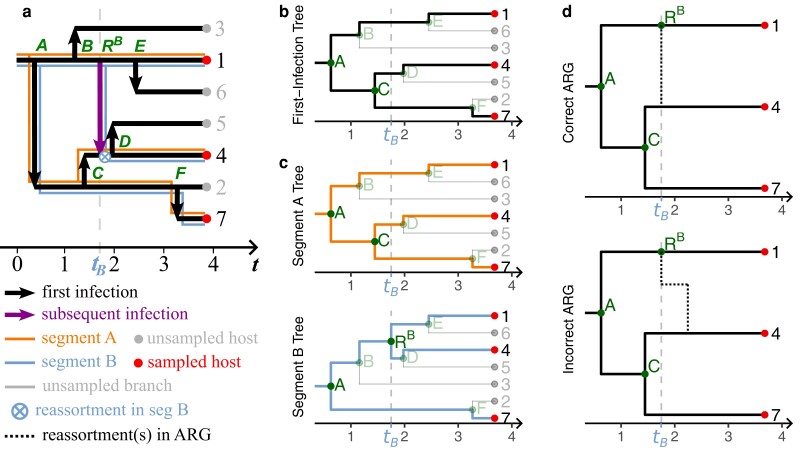
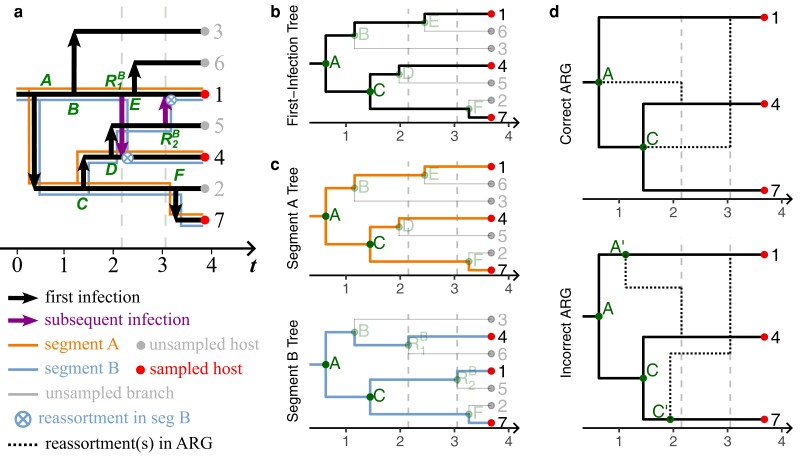
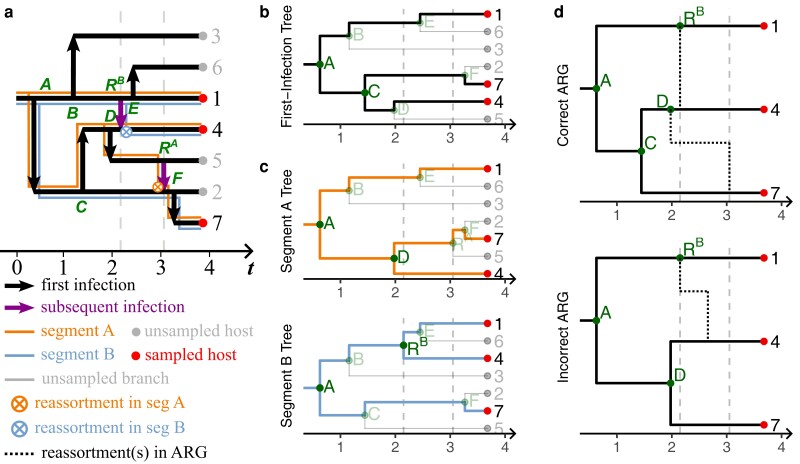
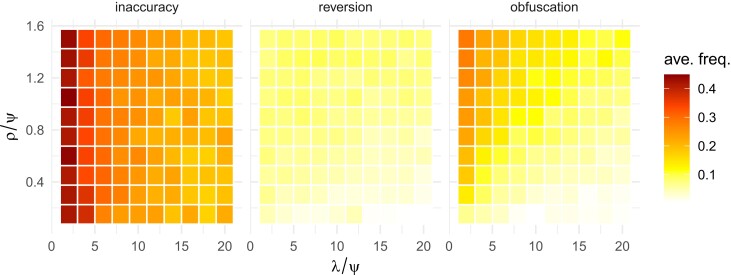
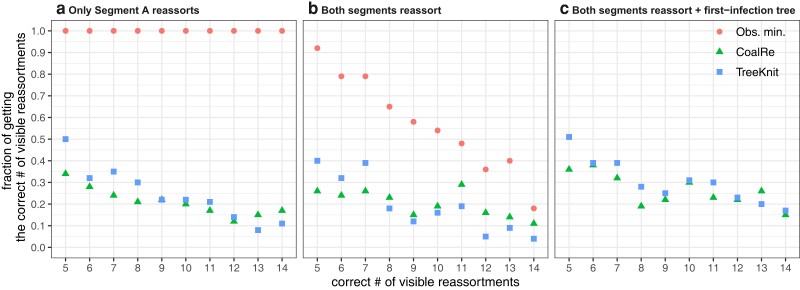
Reassortment is an evolutionary process common in viruses with segmented genomes. These viruses can swap whole genomic segments during cellular co-infection, giving rise to novel progeny formed from the mixture of parental segments. Since large-scale genome rearrangements have the potential to generate new phenotypes, reassortment is important to both evolutionary biology and public health research. However, statistical inference of the pattern of reassortment events from phylogenetic data is exceptionally difficult, potentially involving inference of general graphs in which individual segment trees are embedded. In this paper, we argue that, in general, the number and pattern of reassortment events are not identifiable from segment trees alone, even with theoretically ideal data. We call this fact the fundamental problem of reassortment, which we illustrate using the concept of the "first-infection tree," a potentially counterfactual genealogy that would have been observed in the segment trees had no reassortment occurred. Further, we illustrate four additional problems that can arise logically in the inference of reassortment events and show, using simulated data, that these problems are not rare and can potentially distort our observation of reassortment even in small data sets. Finally, we discuss how existing methods can be augmented or adapted to account for not only the fundamental problem of reassortment, but also the four additional situations that can complicate the inference of reassortment.
Keywords: genomic reassortment; molecular epidemiology; phylodynamics; population genetics.
© The Author(s) 2024. Published by Oxford University Press on behalf of Society for Molecular Biology and Evolution.
Conflict of interest statement
Conflict of Interest None declared.
Figures
Similar articles
-
A phylogenetic approach to detecting reassortments in viruses with segmented genomes.Gene. 2010 Sep 15;464(1-2):11-6. doi: 10.1016/j.gene.2010.05.002. Epub 2010 May 28. Gene. 2010. PMID: 20546849
-
Modeling the evolution of segment trees reveals deficiencies in current inferential methods for genomic reassortment.bioRxiv [Preprint]. 2023 Sep 22:2023.09.20.558687. doi: 10.1101/2023.09.20.558687. bioRxiv. 2023. PMID: 37790507 Free PMC article. Preprint.
-
Bayesian inference of reassortment networks reveals fitness benefits of reassortment in human influenza viruses.Proc Natl Acad Sci U S A. 2020 Jul 21;117(29):17104-17111. doi: 10.1073/pnas.1918304117. Epub 2020 Jul 6. Proc Natl Acad Sci U S A. 2020. PMID: 32631984 Free PMC article.
-
Progress and Challenge in Computational Identification of Influenza Virus Reassortment.Virol Sin. 2021 Dec;36(6):1273-1283. doi: 10.1007/s12250-021-00392-w. Epub 2021 May 26. Virol Sin. 2021. PMID: 34037948 Free PMC article. Review.
-
Implications of segment mismatch for influenza A virus evolution.J Gen Virol. 2018 Jan;99(1):3-16. doi: 10.1099/jgv.0.000989. Epub 2017 Dec 15. J Gen Virol. 2018. PMID: 29244017 Free PMC article. Review.
Cited by
-
Isolation and characterization of a novel coltivirus from Haemaphysalis concinna ticks in Northeastern China.Infect Med (Beijing). 2025 Apr 30;4(2):100179. doi: 10.1016/j.imj.2025.100179. eCollection 2025 Jun. Infect Med (Beijing). 2025. PMID: 40511403 Free PMC article.
References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
