

Targeted accurate RNA consensus sequencing (tARC-seq) reveals mechanisms of replication error affecting SARS-CoV-2 divergence
- PMID: 38649410
- PMCID: PMC11384275
- DOI: 10.1038/s41564-024-01655-4
Targeted accurate RNA consensus sequencing (tARC-seq) reveals mechanisms of replication error affecting SARS-CoV-2 divergence
Abstract
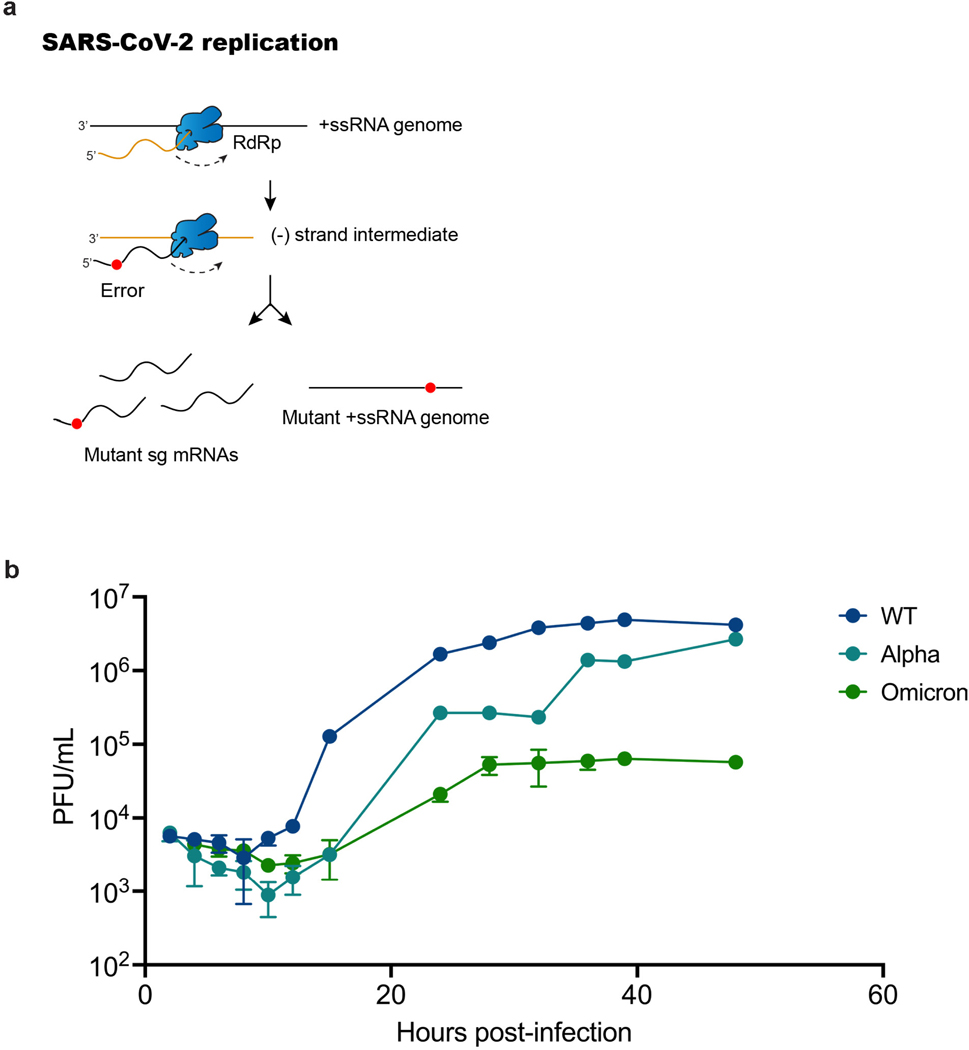
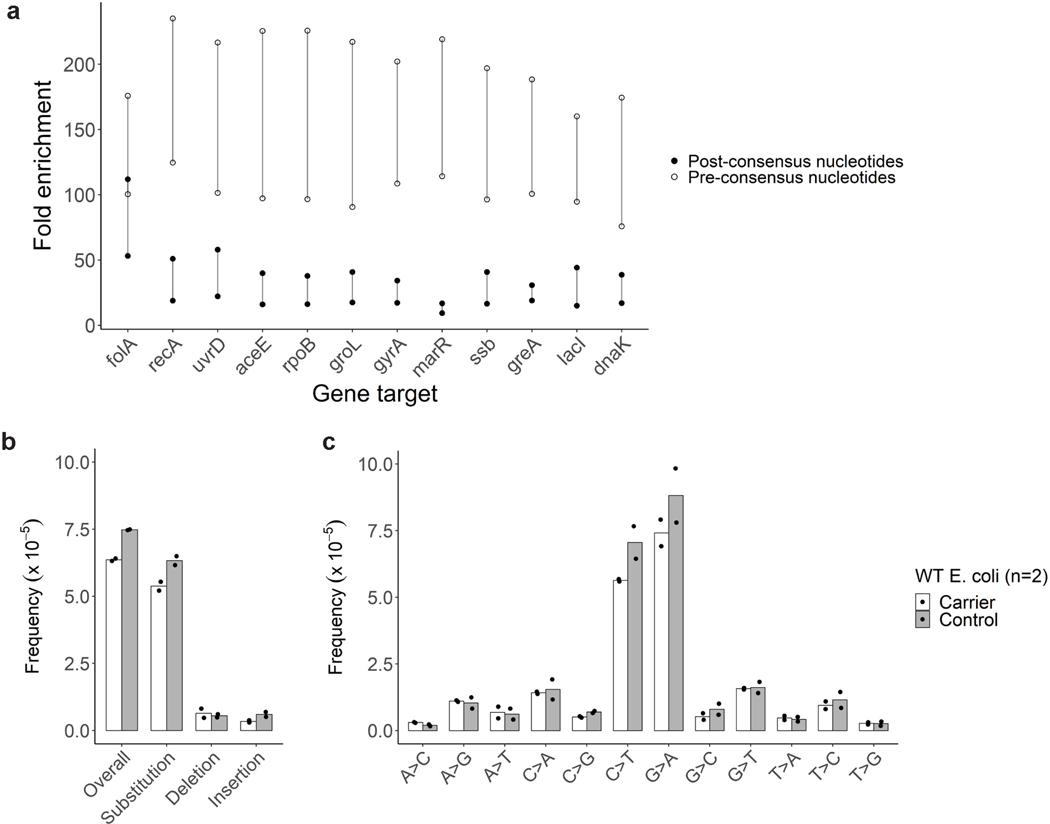
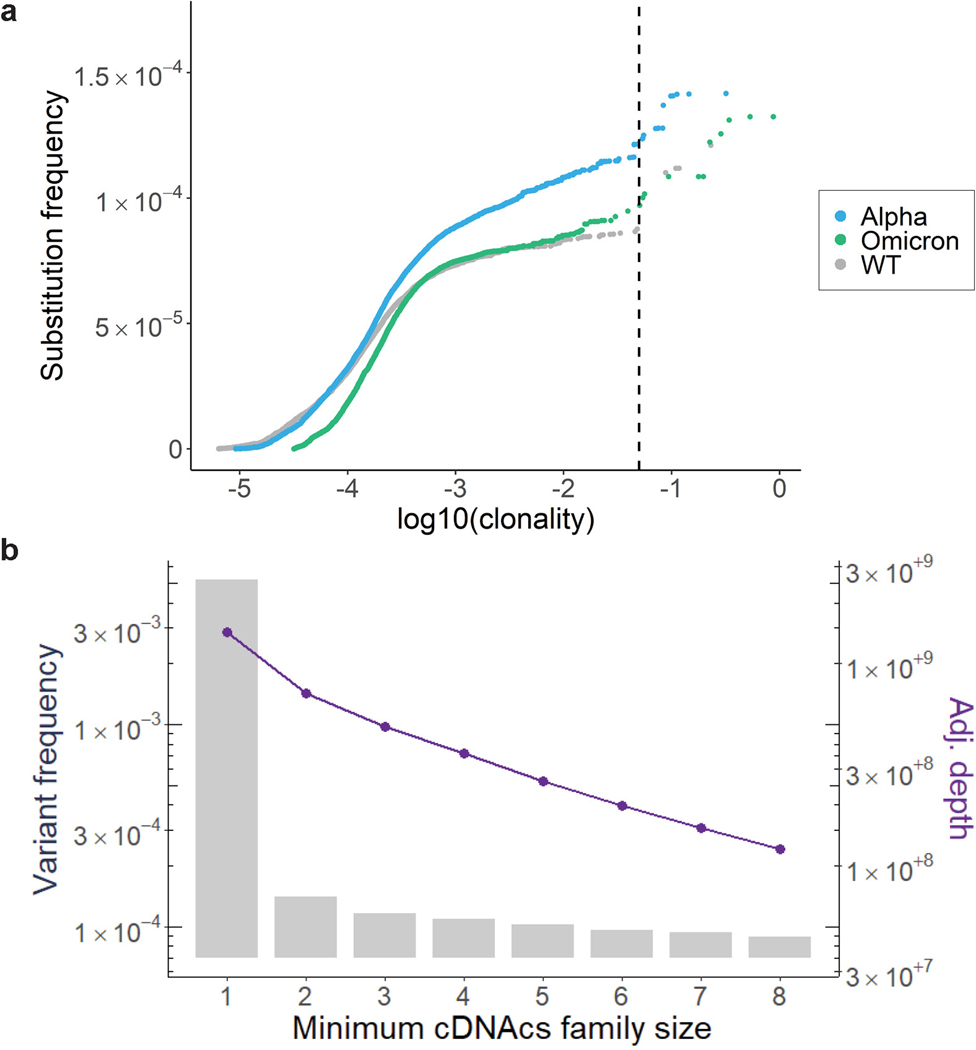
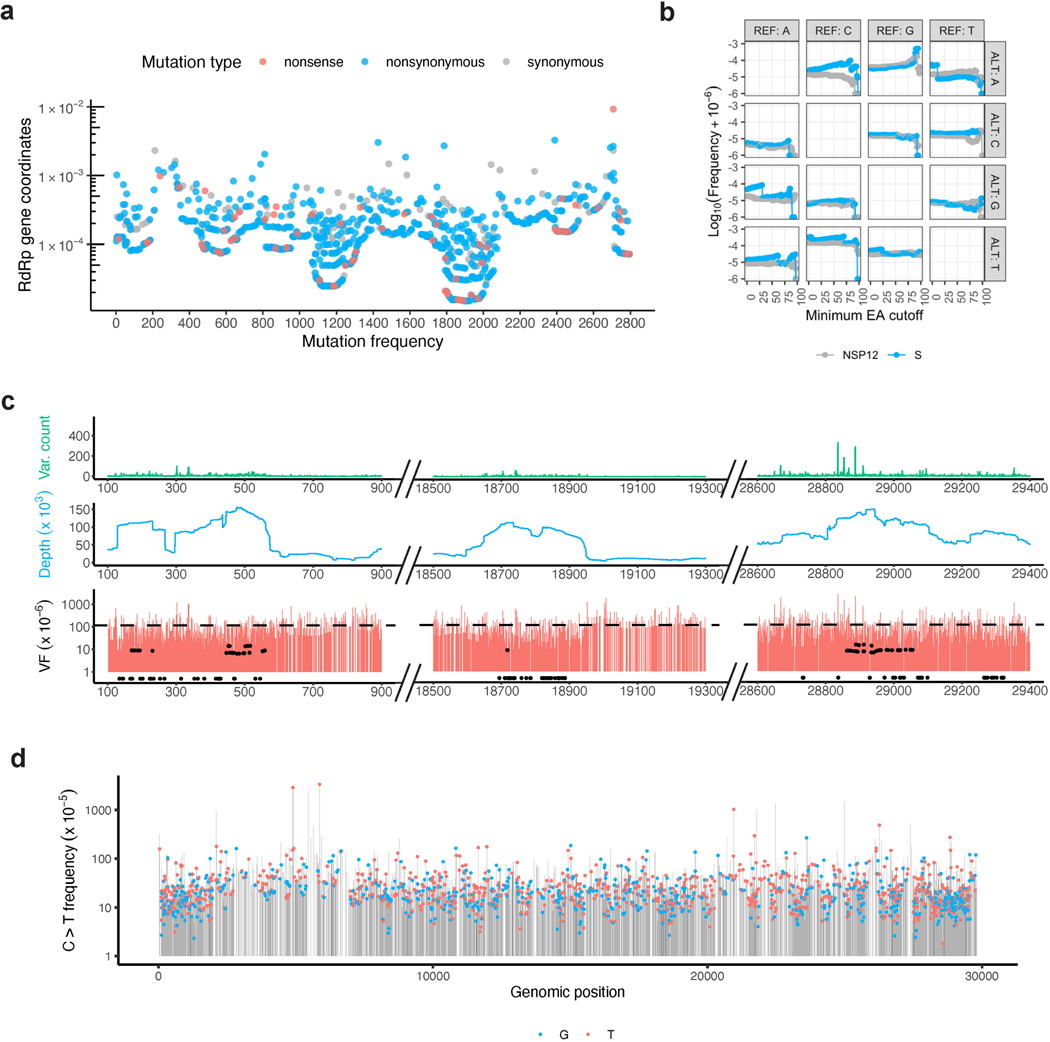
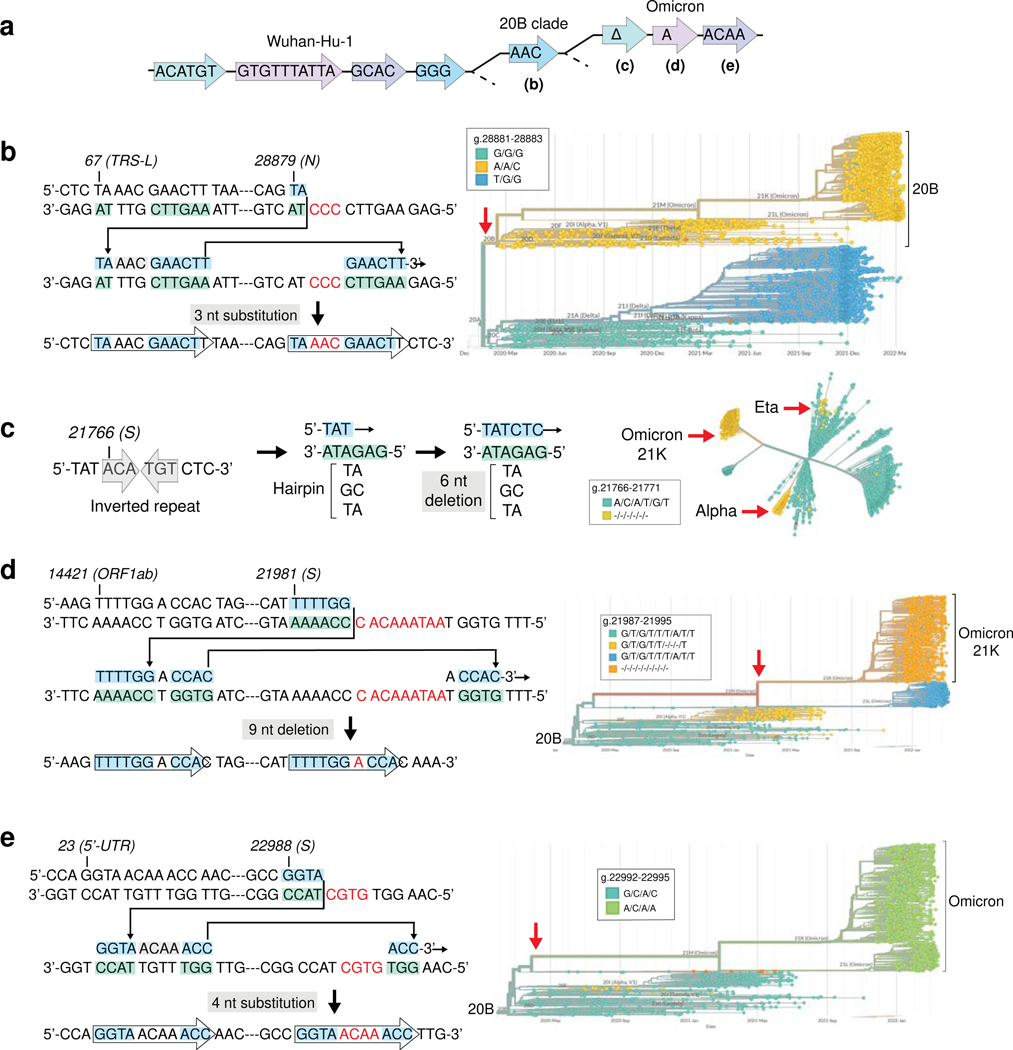
RNA viruses, like SARS-CoV-2, depend on their RNA-dependent RNA polymerases (RdRp) for replication, which is error prone. Monitoring replication errors is crucial for understanding the virus's evolution. Current methods lack the precision to detect rare de novo RNA mutations, particularly in low-input samples such as those from patients. Here we introduce a targeted accurate RNA consensus sequencing method (tARC-seq) to accurately determine the mutation frequency and types in SARS-CoV-2, both in cell culture and clinical samples. Our findings show an average of 2.68 × 10-5 de novo errors per cycle with a C > T bias that cannot be solely attributed to APOBEC editing. We identified hotspots and cold spots throughout the genome, correlating with high or low GC content, and pinpointed transcription regulatory sites as regions more susceptible to errors. tARC-seq captured template switching events including insertions, deletions and complex mutations. These insights shed light on the genetic diversity generation and evolutionary dynamics of SARS-CoV-2.
© 2024. The Author(s), under exclusive licence to Springer Nature Limited.
Conflict of interest statement
Figures

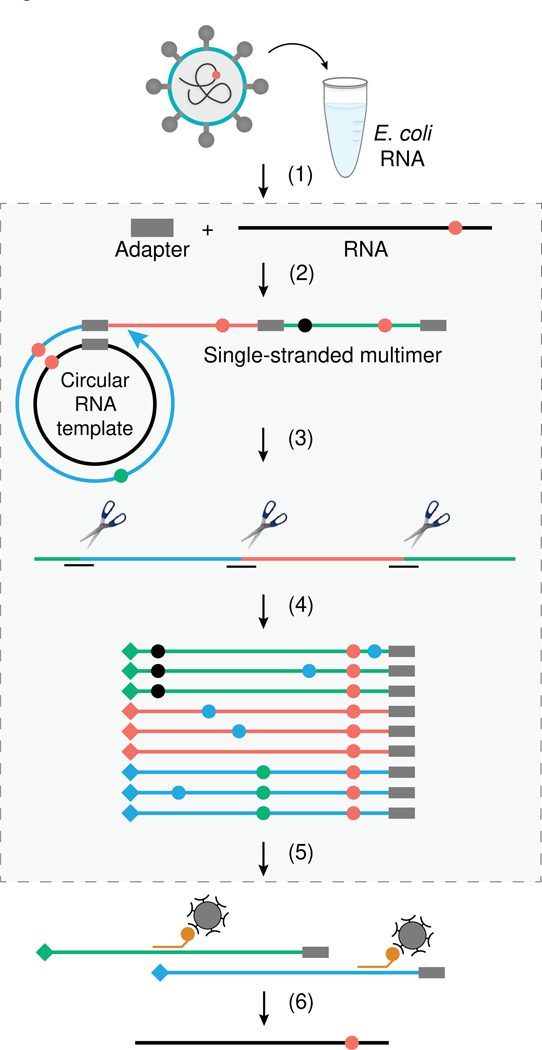
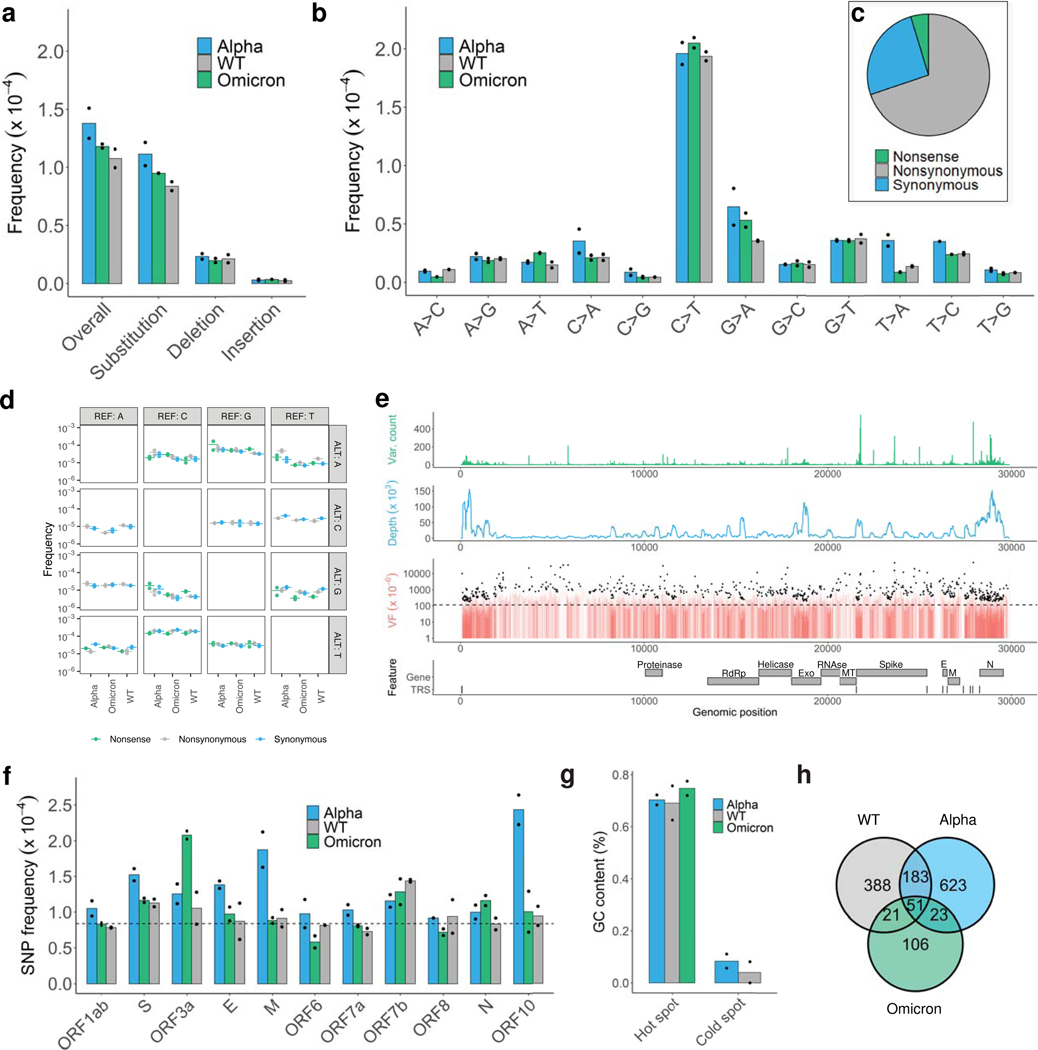
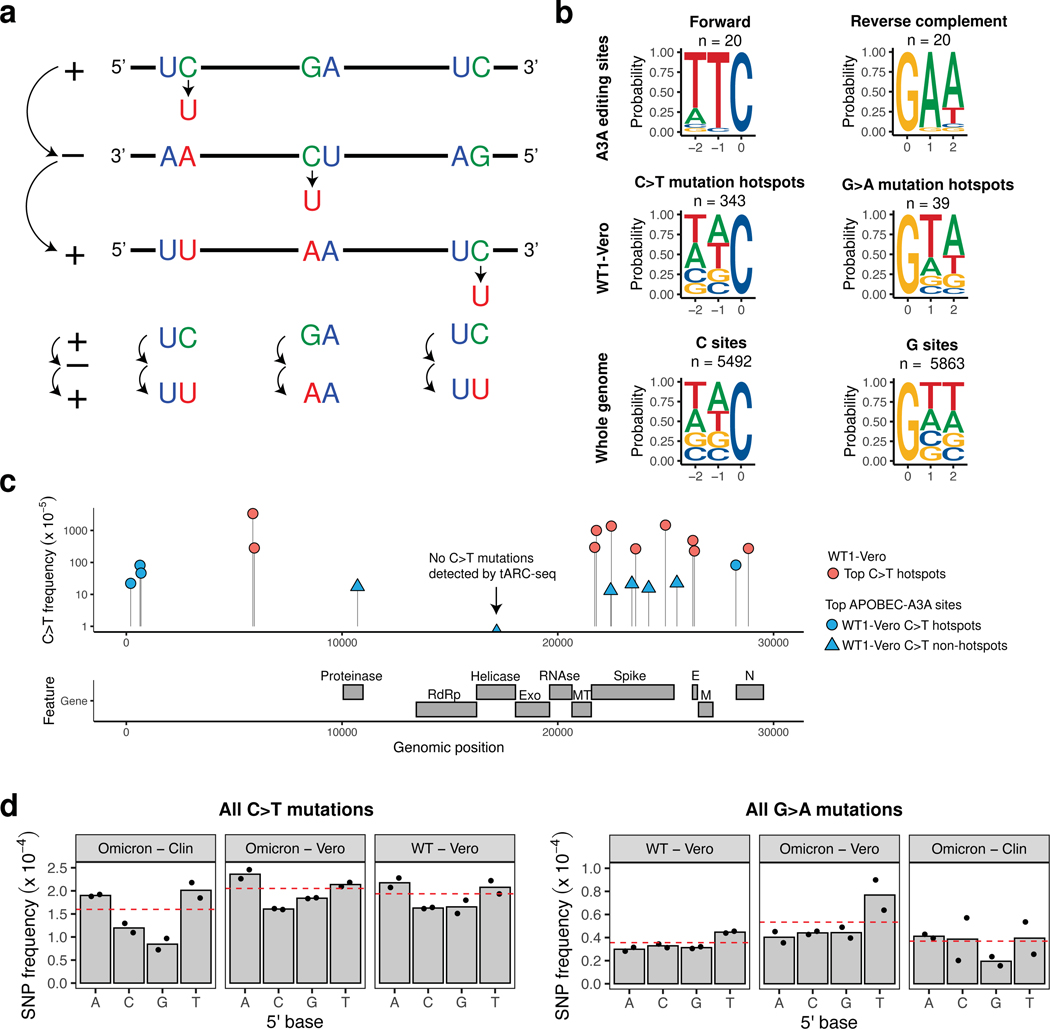
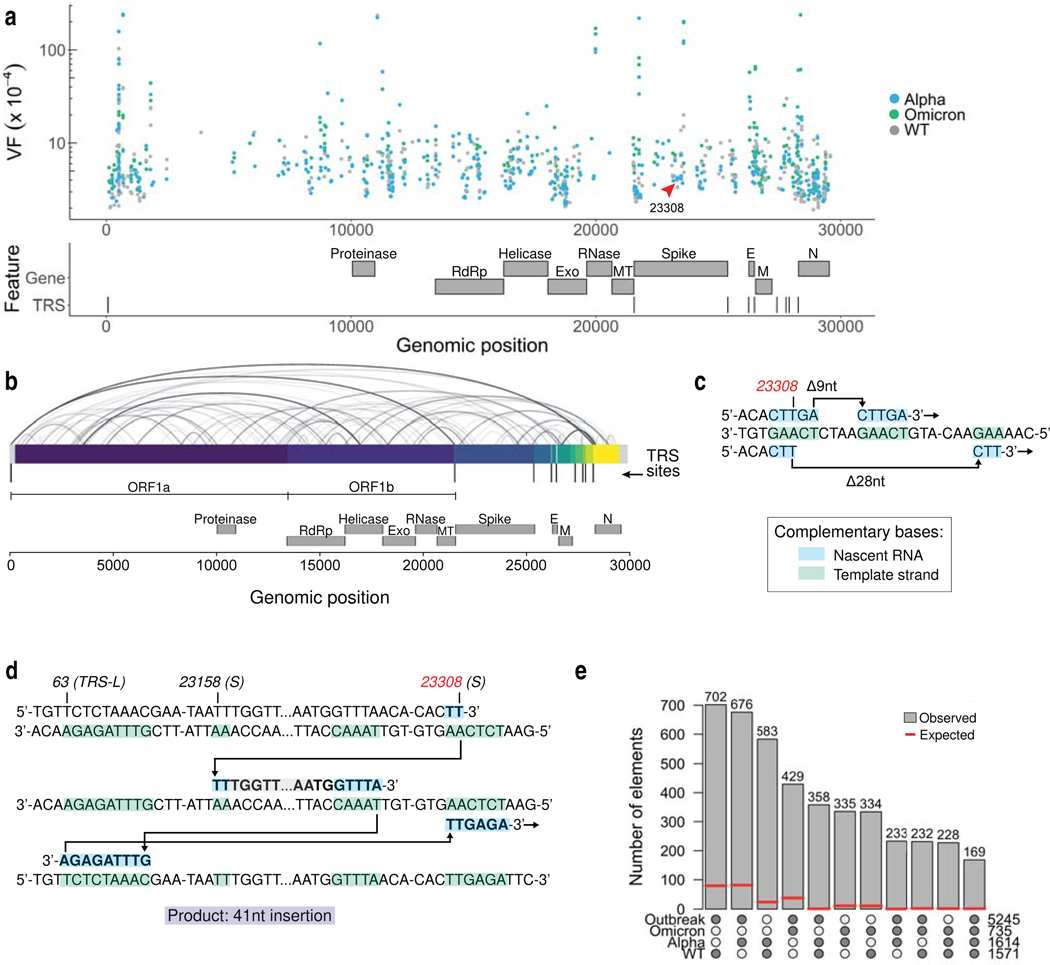
Update of
-
RNA polymerase inaccuracy underlies SARS-CoV-2 variants and vaccine heterogeneity.Res Sq [Preprint]. 2022 Jun 2:rs.3.rs-1690086. doi: 10.21203/rs.3.rs-1690086/v1. Res Sq. 2022. Update in: Nat Microbiol. 2024 May;9(5):1382-1392. doi: 10.1038/s41564-024-01655-4. PMID: 35677076 Free PMC article. Updated. Preprint.
Similar articles
-
RNA polymerase inaccuracy underlies SARS-CoV-2 variants and vaccine heterogeneity.Res Sq [Preprint]. 2022 Jun 2:rs.3.rs-1690086. doi: 10.21203/rs.3.rs-1690086/v1. Res Sq. 2022. Update in: Nat Microbiol. 2024 May;9(5):1382-1392. doi: 10.1038/s41564-024-01655-4. PMID: 35677076 Free PMC article. Updated. Preprint.
-
Profiling SARS-CoV-2 mutation fingerprints that range from the viral pangenome to individual infection quasispecies.Genome Med. 2021 Apr 19;13(1):62. doi: 10.1186/s13073-021-00882-2. Genome Med. 2021. PMID: 33875001 Free PMC article.
-
Host-Virus Chimeric Events in SARS-CoV-2-Infected Cells Are Infrequent and Artifactual.J Virol. 2021 Jul 12;95(15):e0029421. doi: 10.1128/JVI.00294-21. Epub 2021 Jul 12. J Virol. 2021. PMID: 33980601 Free PMC article.
-
From RNA World to SARS-CoV-2: The Edited Story of RNA Viral Evolution.Cells. 2021 Jun 20;10(6):1557. doi: 10.3390/cells10061557. Cells. 2021. PMID: 34202997 Free PMC article. Review.
-
A Glimpse on the Evolution of RNA Viruses: Implications and Lessons from SARS-CoV-2.Viruses. 2022 Dec 20;15(1):1. doi: 10.3390/v15010001. Viruses. 2022. PMID: 36680042 Free PMC article. Review.
Cited by
-
Changes in Phenotypic and Molecular Features of Naïve and Central Memory T Helper Cell Subsets following SARS-CoV-2 Vaccination.Vaccines (Basel). 2024 Sep 11;12(9):1040. doi: 10.3390/vaccines12091040. Vaccines (Basel). 2024. PMID: 39340069 Free PMC article.
-
SARS-CoV-2 resistance analyses from the Phase 3 PINETREE study of remdesivir treatment in nonhospitalized participants.Antimicrob Agents Chemother. 2025 Feb 13;69(2):e0123824. doi: 10.1128/aac.01238-24. Epub 2024 Dec 19. Antimicrob Agents Chemother. 2025. PMID: 39699245 Free PMC article. Clinical Trial.
-
The mutational landscape of SARS-CoV-2 provides new insight into viral evolution and fitness.Nat Commun. 2025 Jul 11;16(1):6425. doi: 10.1038/s41467-025-61555-x. Nat Commun. 2025. PMID: 40645940 Free PMC article.
-
Refining SARS-CoV-2 intra-host variation by leveraging large-scale sequencing data.NAR Genom Bioinform. 2024 Nov 12;6(4):lqae145. doi: 10.1093/nargab/lqae145. eCollection 2024 Sep. NAR Genom Bioinform. 2024. PMID: 39534500 Free PMC article.
-
APOBEC3-related mutations in the spike protein-encoding region facilitate SARS-CoV-2 evolution.Heliyon. 2024 May 29;10(11):e32139. doi: 10.1016/j.heliyon.2024.e32139. eCollection 2024 Jun 15. Heliyon. 2024. PMID: 38868014 Free PMC article.
References
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
- R01GM088653/U.S. Department of Health & Human Services | NIH | National Institute of General Medical Sciences (NIGMS)
- 1R21CA259780/U.S. Department of Health & Human Services | NIH | National Cancer Institute (NCI)
- 1R21HG011229/U.S. Department of Health & Human Services | National Institutes of Health (NIH)
- DBI-2032904/National Science Foundation (NSF)
- 3R01AG061105-03S1/U.S. Department of Health & Human Services | NIH | National Institute on Aging (U.S. National Institute on Aging)
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
