

Updated mechanisms of MASLD pathogenesis
- PMID: 38649999
- PMCID: PMC11034170
- DOI: 10.1186/s12944-024-02108-x
Updated mechanisms of MASLD pathogenesis
Abstract
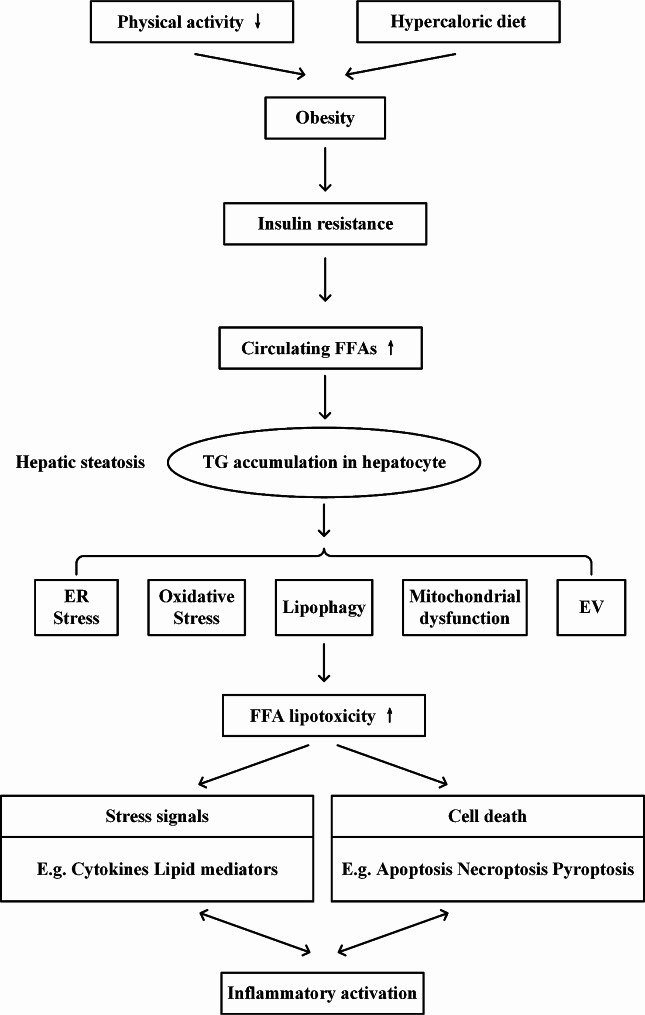
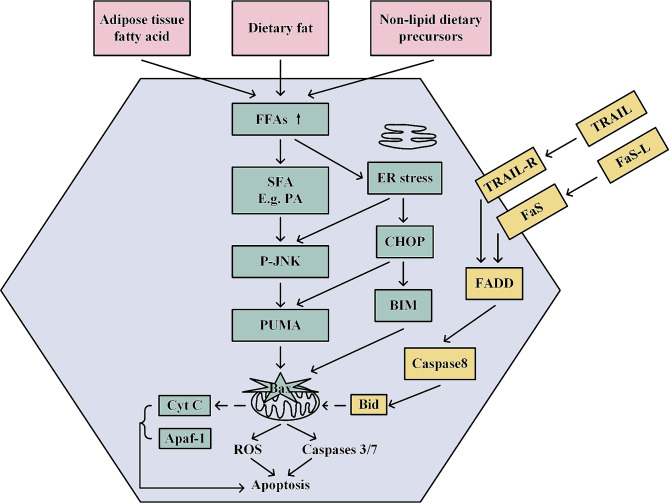
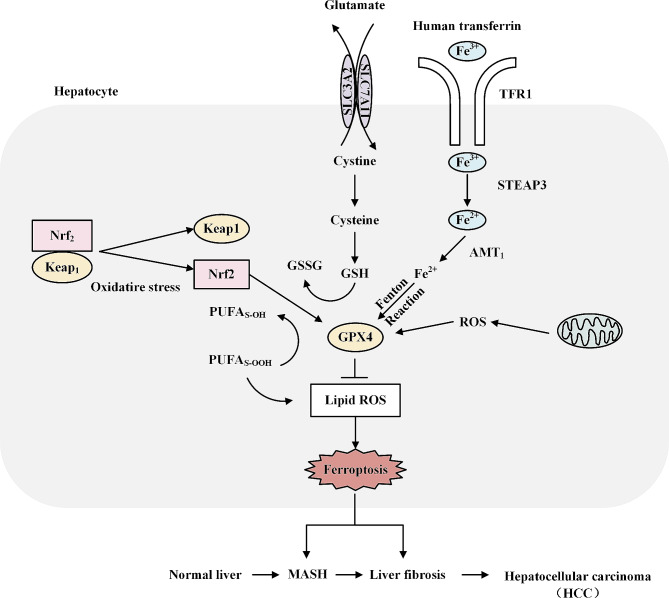
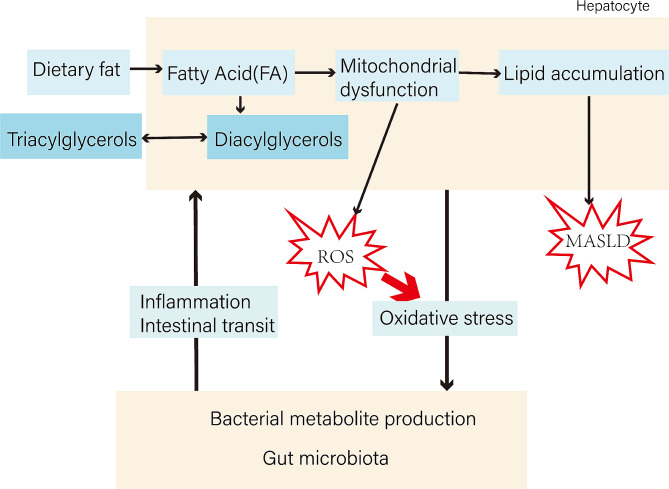
Metabolic dysfunction-associated steatotic liver disease (MASLD) has garnered considerable attention globally. Changing lifestyles, over-nutrition, and physical inactivity have promoted its development. MASLD is typically accompanied by obesity and is strongly linked to metabolic syndromes. Given that MASLD prevalence is on the rise, there is an urgent need to elucidate its pathogenesis. Hepatic lipid accumulation generally triggers lipotoxicity and induces MASLD or progress to metabolic dysfunction-associated steatohepatitis (MASH) by mediating endoplasmic reticulum stress, oxidative stress, organelle dysfunction, and ferroptosis. Recently, significant attention has been directed towards exploring the role of gut microbial dysbiosis in the development of MASLD, offering a novel therapeutic target for MASLD. Considering that there are no recognized pharmacological therapies due to the diversity of mechanisms involved in MASLD and the difficulty associated with undertaking clinical trials, potential targets in MASLD remain elusive. Thus, this article aimed to summarize and evaluate the prominent roles of lipotoxicity, ferroptosis, and gut microbes in the development of MASLD and the mechanisms underlying their effects. Furthermore, existing advances and challenges in the treatment of MASLD were outlined.
Keywords: Lipid metabolism; Lipotoxicity; MASLD; Therapeutics.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
