

Allelic heterogeneity and abnormal vesicle recycling in PLAA-related neurodevelopmental disorders
- PMID: 38650658
- PMCID: PMC11033462
- DOI: 10.3389/fnmol.2024.1268013
Allelic heterogeneity and abnormal vesicle recycling in PLAA-related neurodevelopmental disorders
Abstract
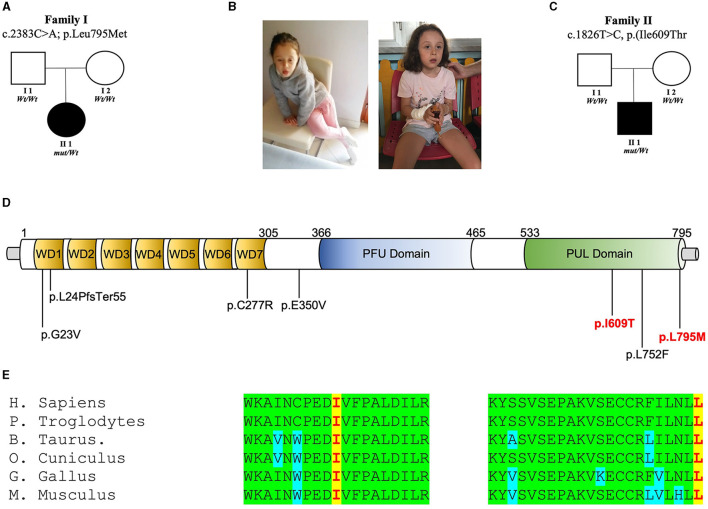
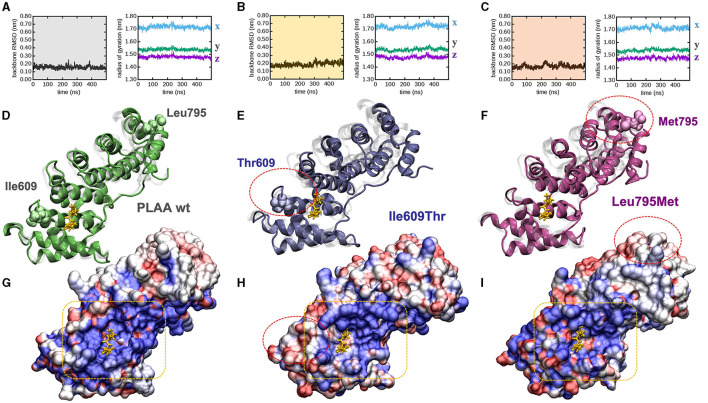
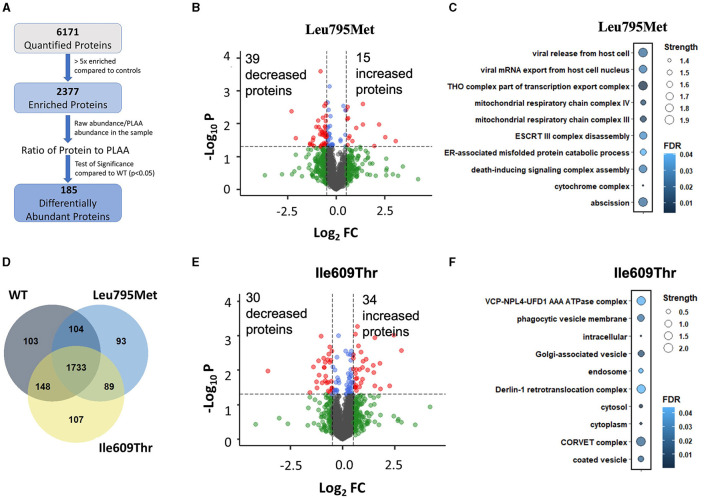
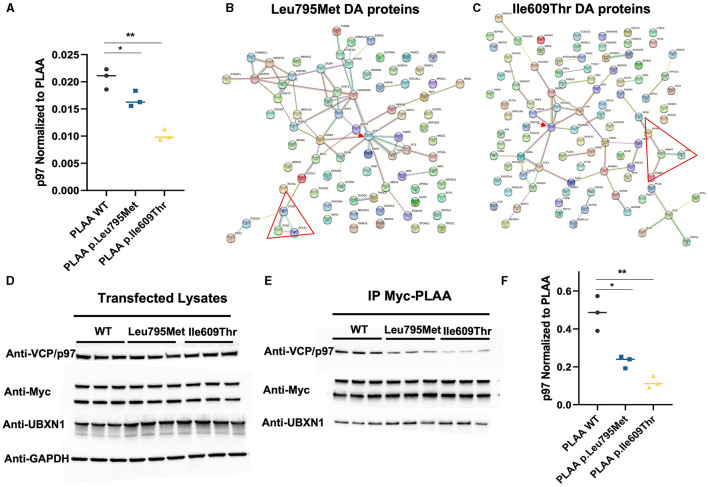
The human PLAA gene encodes Phospholipase-A2-Activating-Protein (PLAA) involved in trafficking of membrane proteins. Through its PUL domain (PLAP, Ufd3p, and Lub1p), PLAA interacts with p97/VCP modulating synaptic vesicles recycling. Although few families carrying biallelic PLAA variants were reported with progressive neurodegeneration, consequences of monoallelic PLAA variants have not been elucidated. Using exome or genome sequencing we identified PLAA de-novo missense variants, affecting conserved residues within the PUL domain, in children affected with neurodevelopmental disorders (NDDs), including psychomotor regression, intellectual disability (ID) and autism spectrum disorders (ASDs). Computational and in-vitro studies of the identified variants revealed abnormal chain arrangements at C-terminal and reduced PLAA-p97/VCP interaction, respectively. These findings expand both allelic and phenotypic heterogeneity associated to PLAA-related neurological disorders, highlighting perturbed vesicle recycling as a potential disease mechanism in NDDs due to genetic defects of PLAA.
Keywords: PLAA gene; SNAREopathies; de novo variants; developmental regression; neurodevelopmental disorders; synaptic transmission.
Copyright © 2024 Iacomino, Houerbi, Fortuna, Howe, Li, Scorrano, Riva, Cheng, Steiman, Peltekova, Yusuf, Baldassari, Tamburro, Scudieri, Musante, Di Ludovico, Guerrisi, Balagura, Corsello, Efthymiou, Murphy, Uva, Verrotti, Fiorillo, Delvecchio, Accogli, Elsabbagh, Houlden, Scherer, Striano, Zara, Chou and Salpietro.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
-
- American Psychiatric Association (2013). Diagnostic and Statistical Manual of Mental Disorders: DSM-5, 5th Edn. Arlington, VA: American Psychiatric Association.
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous