

Lineage specific transcription factor waves reprogram neuroblastoma from self-renewal to differentiation
- PMID: 38653778
- PMCID: PMC11039666
- DOI: 10.1038/s41467-024-47166-y
Lineage specific transcription factor waves reprogram neuroblastoma from self-renewal to differentiation
Abstract
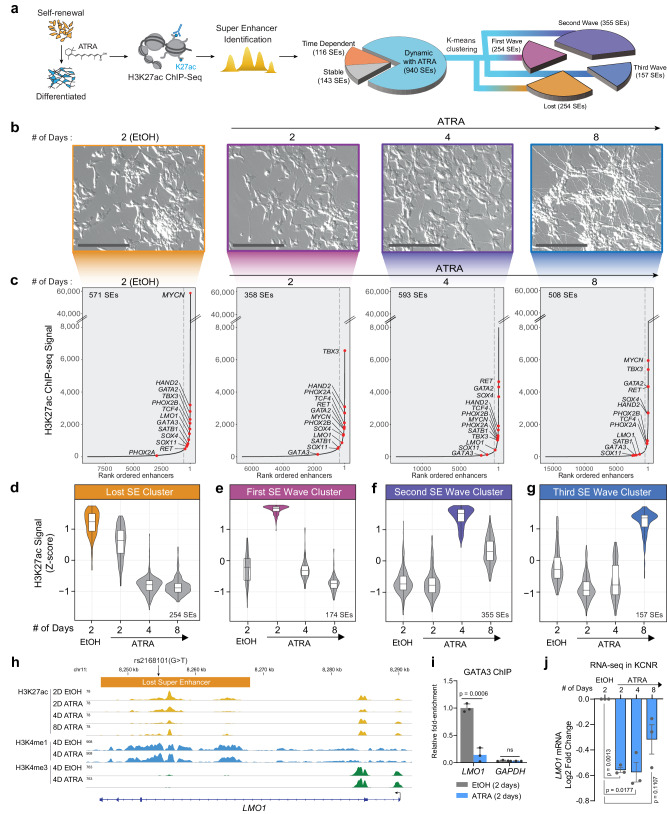
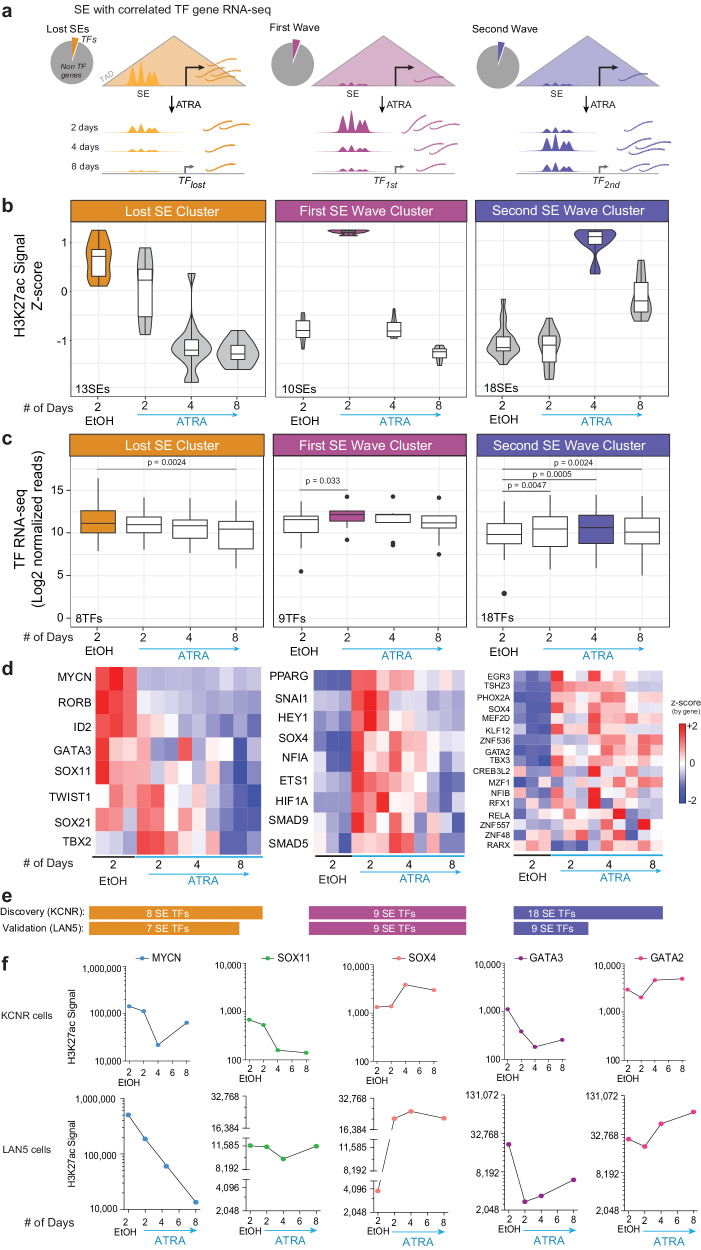
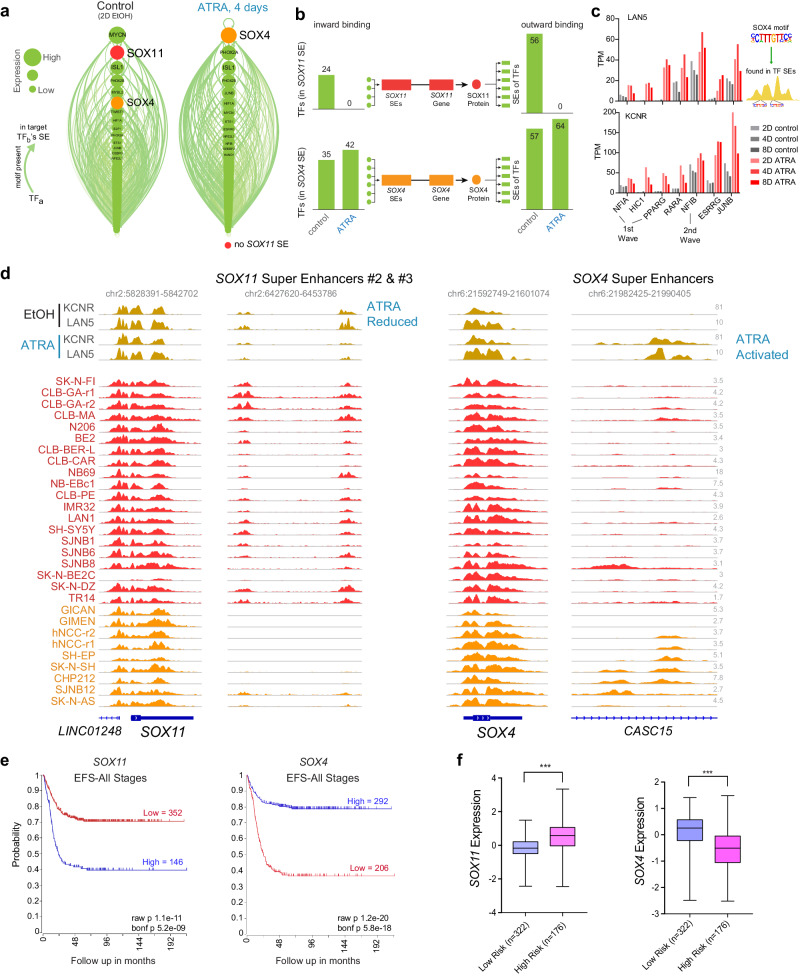
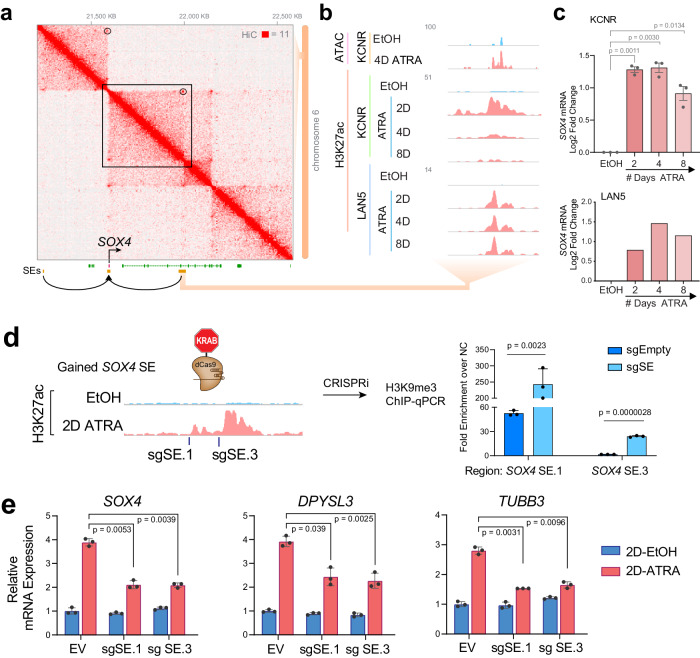
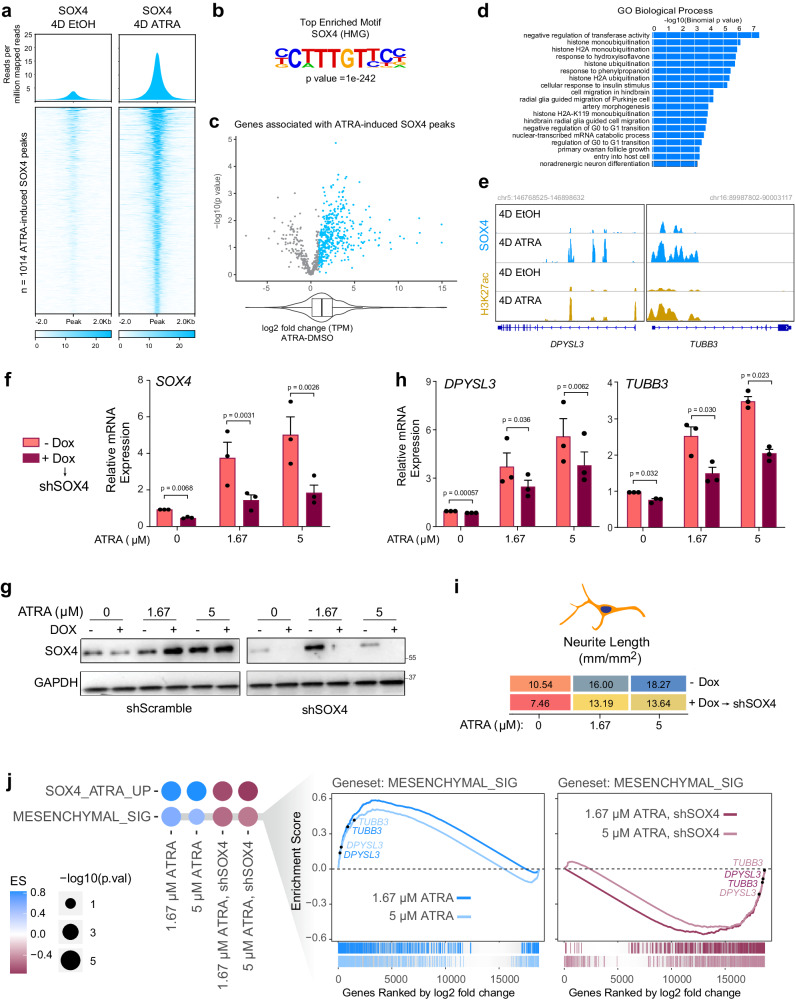
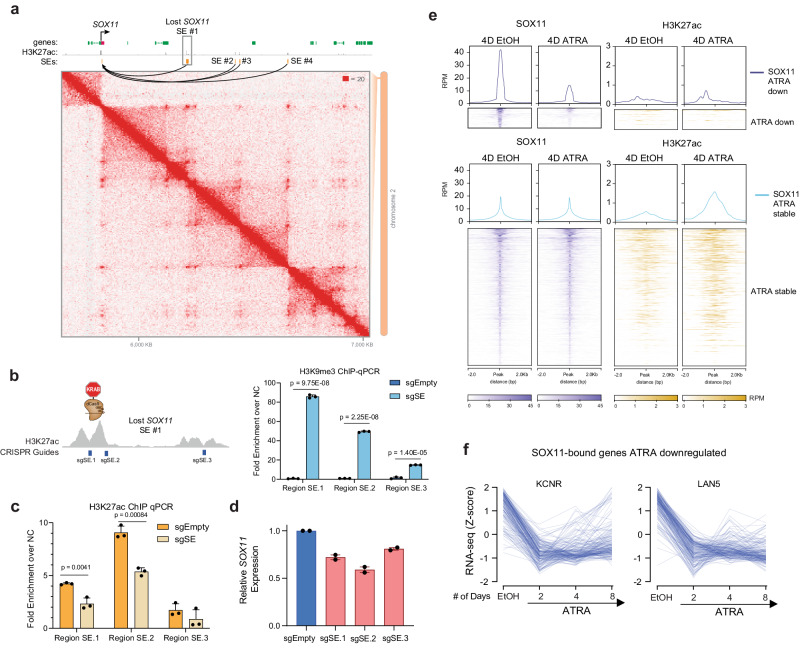
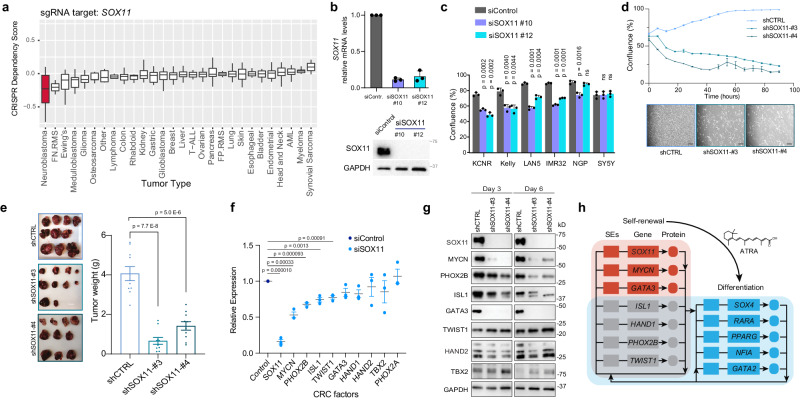
Temporal regulation of super-enhancer (SE) driven transcription factors (TFs) underlies normal developmental programs. Neuroblastoma (NB) arises from an inability of sympathoadrenal progenitors to exit a self-renewal program and terminally differentiate. To identify SEs driving TF regulators, we use all-trans retinoic acid (ATRA) to induce NB growth arrest and differentiation. Time-course H3K27ac ChIP-seq and RNA-seq reveal ATRA coordinated SE waves. SEs that decrease with ATRA link to stem cell development (MYCN, GATA3, SOX11). CRISPR-Cas9 and siRNA verify SOX11 dependency, in vitro and in vivo. Silencing the SOX11 SE using dCAS9-KRAB decreases SOX11 mRNA and inhibits cell growth. Other TFs activate in sequential waves at 2, 4 and 8 days of ATRA treatment that regulate neural development (GATA2 and SOX4). Silencing the gained SOX4 SE using dCAS9-KRAB decreases SOX4 expression and attenuates ATRA-induced differentiation genes. Our study identifies oncogenic lineage drivers of NB self-renewal and TFs critical for implementing a differentiation program.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
