

Undiagnosed RASopathies in infertile men
- PMID: 38654924
- PMCID: PMC11035881
- DOI: 10.3389/fendo.2024.1312357
Undiagnosed RASopathies in infertile men
Abstract
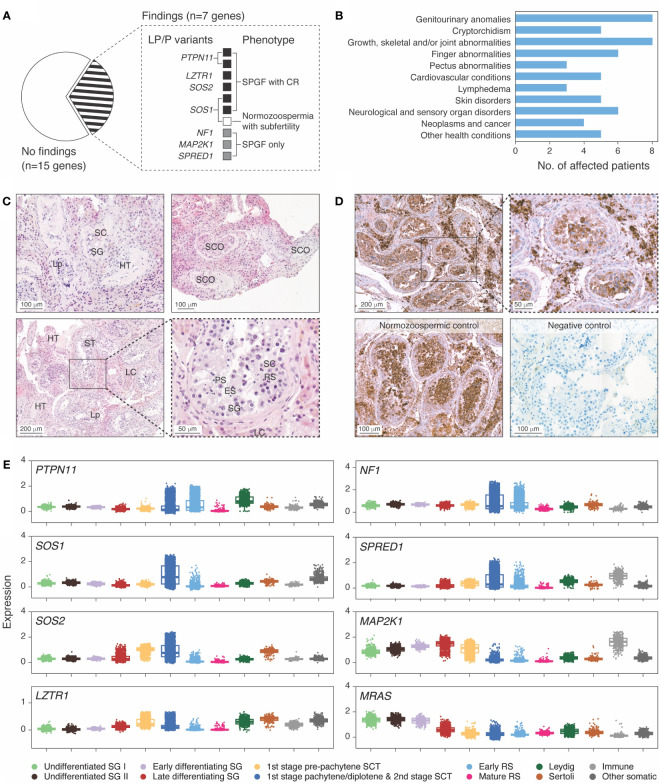
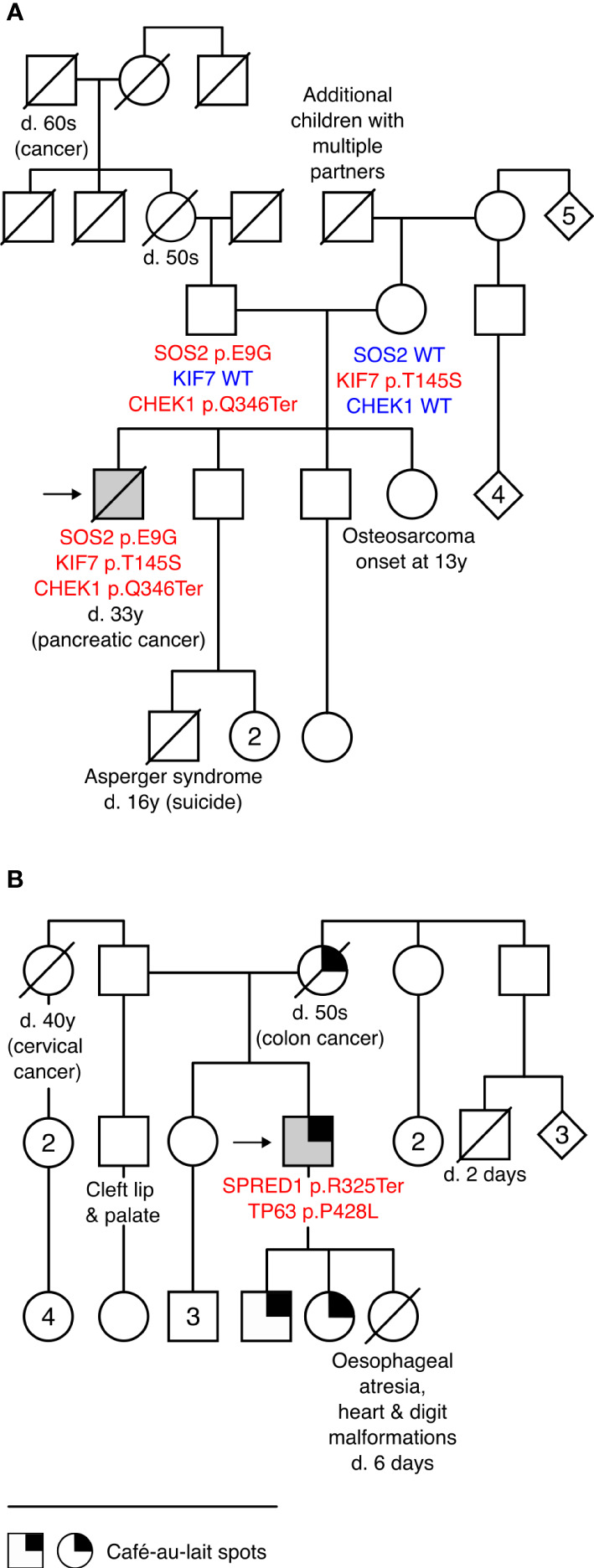
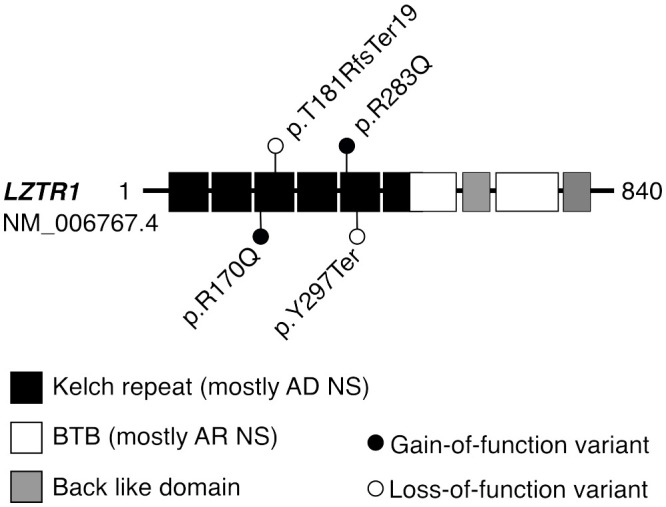
RASopathies are syndromes caused by congenital defects in the Ras/mitogen-activated protein kinase (MAPK) pathway genes, with a population prevalence of 1 in 1,000. Patients are typically identified in childhood based on diverse characteristic features, including cryptorchidism (CR) in >50% of affected men. As CR predisposes to spermatogenic failure (SPGF; total sperm count per ejaculate 0-39 million), we hypothesized that men seeking infertility management include cases with undiagnosed RASopathies. Likely pathogenic or pathogenic (LP/P) variants in 22 RASopathy-linked genes were screened in 521 idiopathic SPGF patients (including 155 CR cases) and 323 normozoospermic controls using exome sequencing. All 844 men were recruited to the ESTonian ANDrology (ESTAND) cohort and underwent identical andrological phenotyping. RASopathy-specific variant interpretation guidelines were used for pathogenicity assessment. LP/P variants were identified in PTPN11 (two), SOS1 (three), SOS2 (one), LZTR1 (one), SPRED1 (one), NF1 (one), and MAP2K1 (one). The findings affected six of 155 cases with CR and SPGF, three of 366 men with SPGF only, and one (of 323) normozoospermic subfertile man. The subgroup "CR and SPGF" had over 13-fold enrichment of findings compared to controls (3.9% vs. 0.3%; Fisher's exact test, p = 5.5 × 10-3). All ESTAND subjects with LP/P variants in the Ras/MAPK pathway genes presented congenital genitourinary anomalies, skeletal and joint conditions, and other RASopathy-linked health concerns. Rare forms of malignancies (schwannomatosis and pancreatic and testicular cancer) were reported on four occasions. The Genetics of Male Infertility Initiative (GEMINI) cohort (1,416 SPGF cases and 317 fertile men) was used to validate the outcome. LP/P variants in PTPN11 (three), LZTR1 (three), and MRAS (one) were identified in six SPGF cases (including 4/31 GEMINI cases with CR) and one normozoospermic man. Undiagnosed RASopathies were detected in total for 17 ESTAND and GEMINI subjects, 15 SPGF patients (10 with CR), and two fertile men. Affected RASopathy genes showed high expression in spermatogenic and testicular somatic cells. In conclusion, congenital defects in the Ras/MAPK pathway genes represent a new congenital etiology of syndromic male infertility. Undiagnosed RASopathies were especially enriched among patients with a history of cryptorchidism. Given the relationship between RASopathies and other conditions, infertile men found to have this molecular diagnosis should be evaluated for known RASopathy-linked health concerns, including specific rare malignancies.
Keywords: RAS/MAPK pathway; cancer; congenital testicular dysgenesis; cryptorchidism; exome sequencing; molecular diagnosis; multidisciplinary management; syndromic male infertility.
Copyright © 2024 Juchnewitsch, Pomm, Dutta, Tamp, Valkna, Lillepea, Mahyari, Tjagur, Belova, Kübarsepp, Castillo-Madeen, Riera-Escamilla, Põlluaas, Nagirnaja, Poolamets, Vihljajev, Sütt, Versbraegen, Papadimitriou, McLachlan, Jarvi, Schlegel, Tennisberg, Korrovits, Vigh-Conrad, O’Bryan, Aston, Lenaerts, Conrad, Kasak, Punab and Laan.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous