

Emergence of enhancers at late DNA replicating regions
- PMID: 38658544
- PMCID: PMC11043393
- DOI: 10.1038/s41467-024-47391-5
Emergence of enhancers at late DNA replicating regions
Abstract
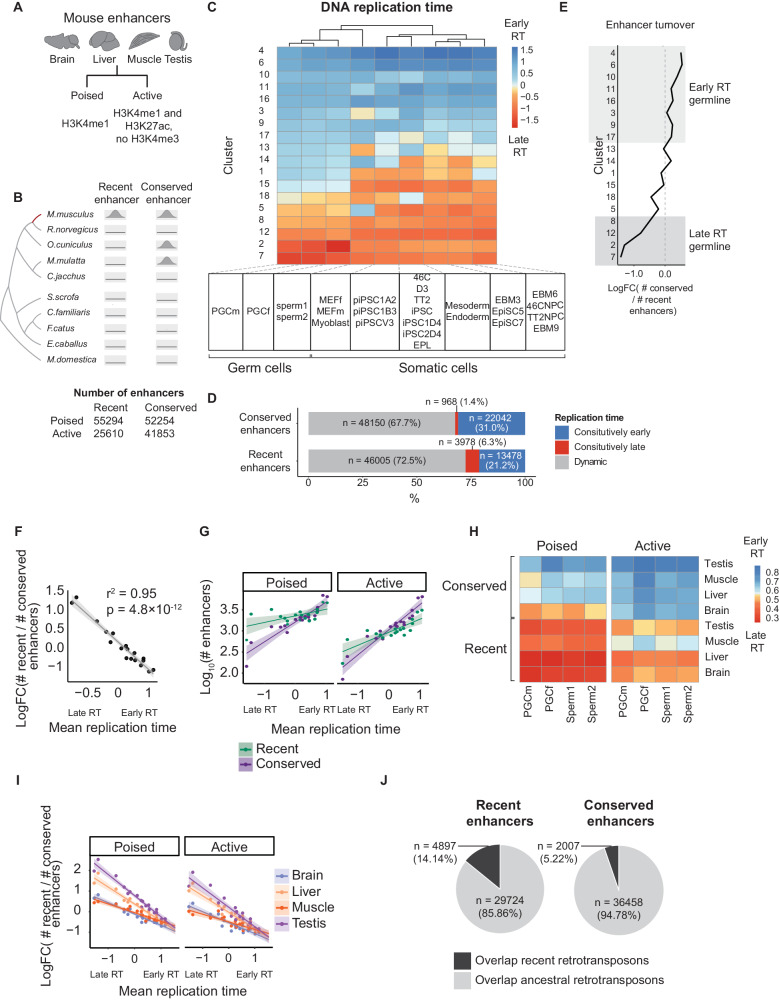
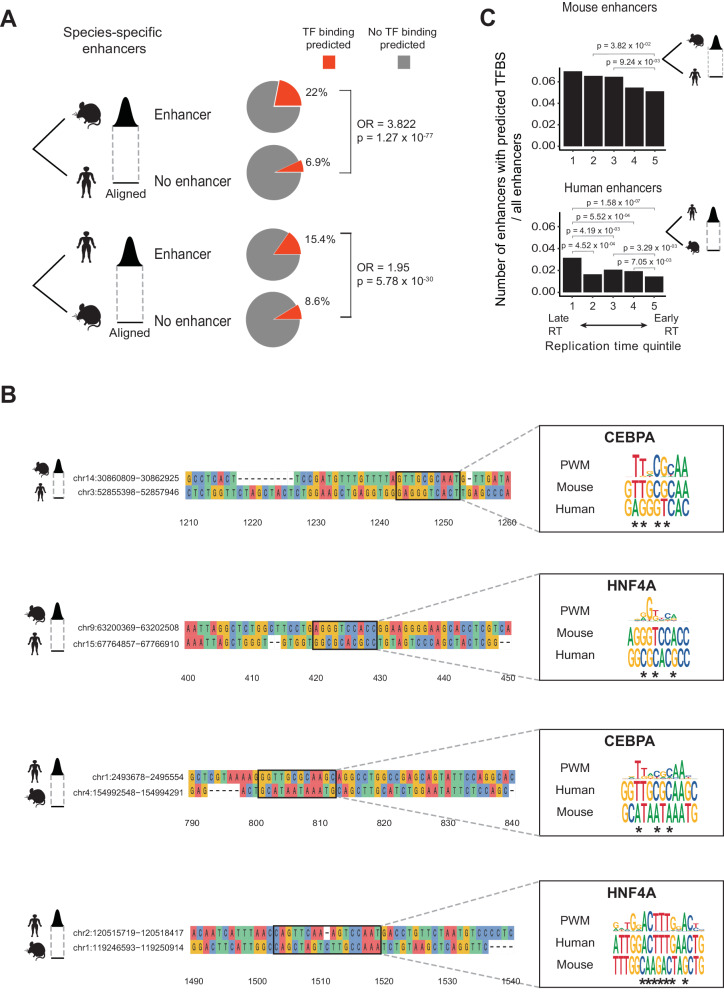
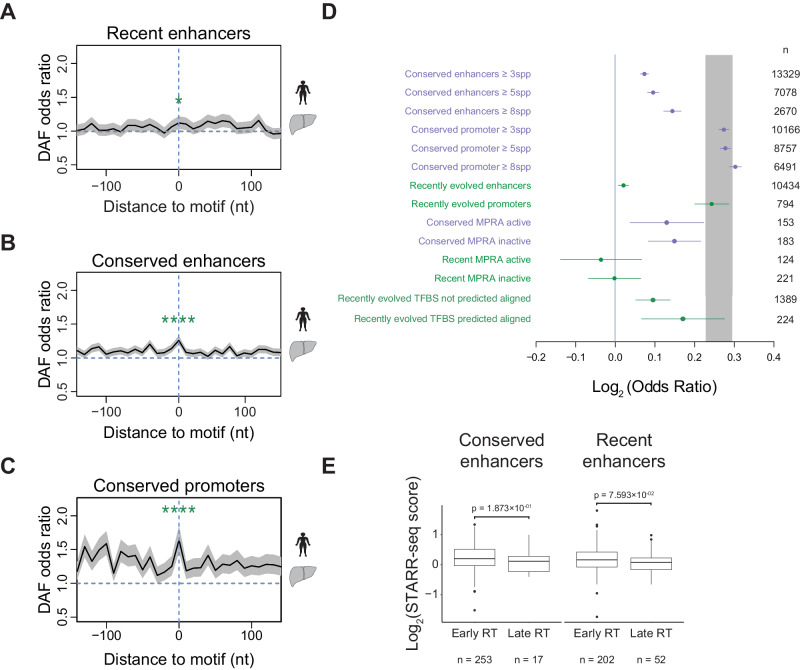
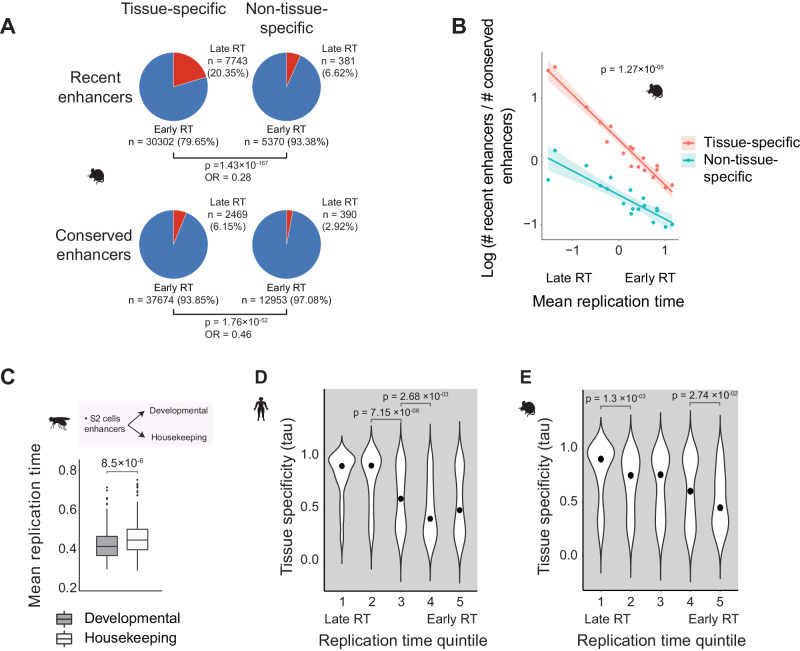
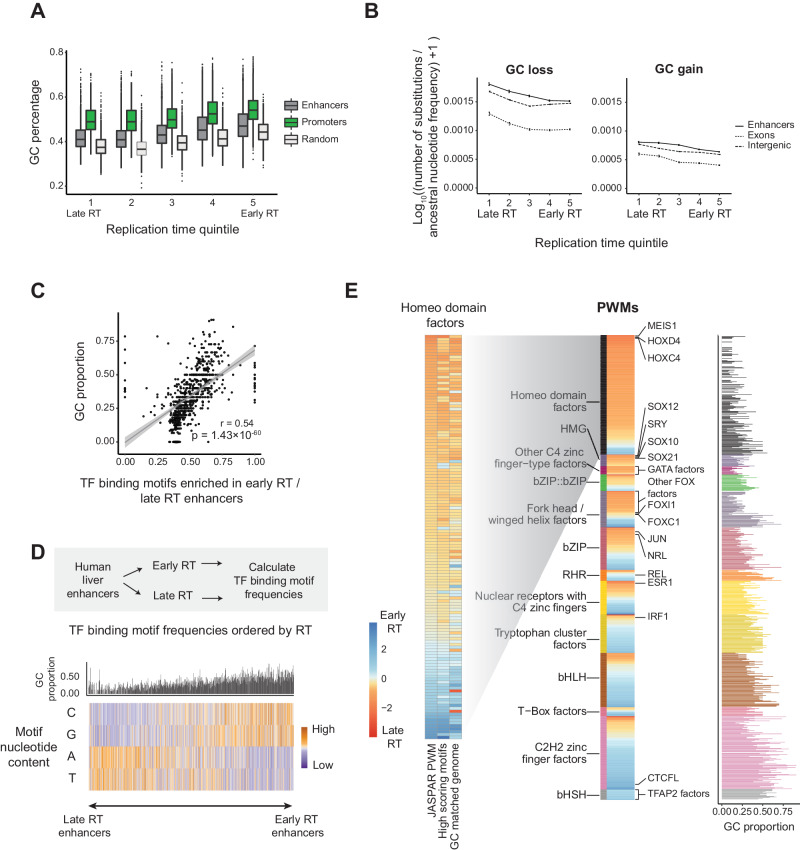
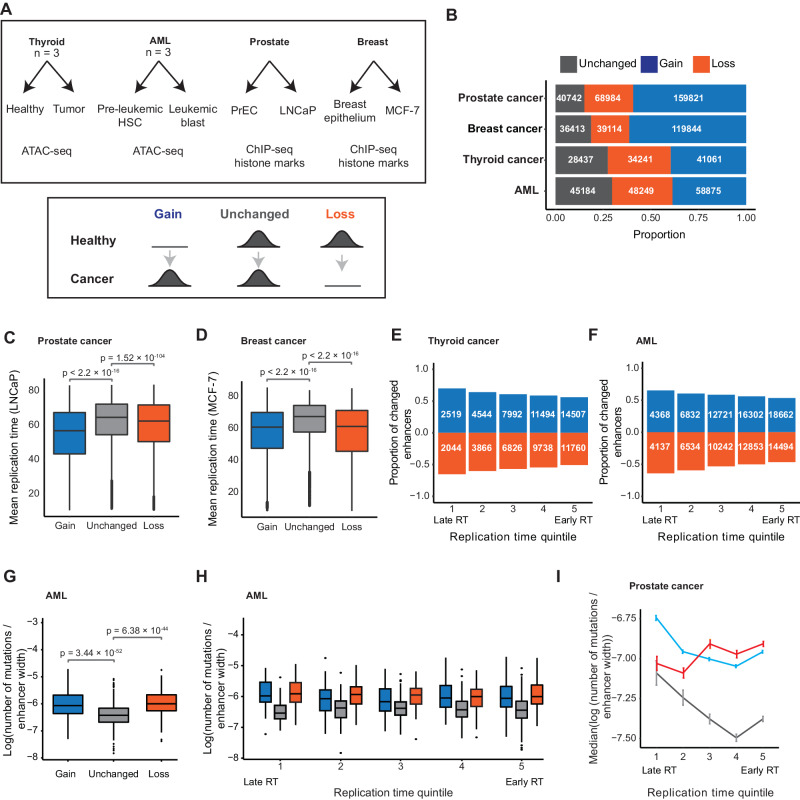
Enhancers are fast-evolving genomic sequences that control spatiotemporal gene expression patterns. By examining enhancer turnover across mammalian species and in multiple tissue types, we uncover a relationship between the emergence of enhancers and genome organization as a function of germline DNA replication time. While enhancers are most abundant in euchromatic regions, enhancers emerge almost twice as often in late compared to early germline replicating regions, independent of transposable elements. Using a deep learning sequence model, we demonstrate that new enhancers are enriched for mutations that alter transcription factor (TF) binding. Recently evolved enhancers appear to be mostly neutrally evolving and enriched in eQTLs. They also show more tissue specificity than conserved enhancers, and the TFs that bind to these elements, as inferred by binding sequences, also show increased tissue-specific gene expression. We find a similar relationship with DNA replication time in cancer, suggesting that these observations may be time-invariant principles of genome evolution. Our work underscores that genome organization has a profound impact in shaping mammalian gene regulation.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
Similar articles
-
Contribution of transposable elements and distal enhancers to evolution of human-specific features of interphase chromatin architecture in embryonic stem cells.Chromosome Res. 2018 Mar;26(1-2):61-84. doi: 10.1007/s10577-018-9571-6. Epub 2018 Jan 15. Chromosome Res. 2018. PMID: 29335803
-
Characterization of sequence determinants of enhancer function using natural genetic variation.Elife. 2022 Aug 31;11:e76500. doi: 10.7554/eLife.76500. Elife. 2022. PMID: 36043696 Free PMC article.
-
Specific subfamilies of transposable elements contribute to different domains of T lymphocyte enhancers.Proc Natl Acad Sci U S A. 2020 Apr 7;117(14):7905-7916. doi: 10.1073/pnas.1912008117. Epub 2020 Mar 19. Proc Natl Acad Sci U S A. 2020. PMID: 32193341 Free PMC article.
-
Actors with Multiple Roles: Pleiotropic Enhancers and the Paradigm of Enhancer Modularity.Trends Genet. 2019 Jun;35(6):423-433. doi: 10.1016/j.tig.2019.03.006. Epub 2019 Apr 18. Trends Genet. 2019. PMID: 31005339 Review.
-
Functional anatomy of distant-acting mammalian enhancers.Philos Trans R Soc Lond B Biol Sci. 2013 May 6;368(1620):20120359. doi: 10.1098/rstb.2012.0359. Print 2013. Philos Trans R Soc Lond B Biol Sci. 2013. PMID: 23650633 Free PMC article. Review.
Cited by
-
Insights into the evolution and regulation of miRNAs from the view of their DNA replication temporal domains.Front Genet. 2025 Jun 23;16:1544802. doi: 10.3389/fgene.2025.1544802. eCollection 2025. Front Genet. 2025. PMID: 40626175 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
