

RAS mutations in myeloid malignancies: revisiting old questions with novel insights and therapeutic perspectives
- PMID: 38658558
- PMCID: PMC11043080
- DOI: 10.1038/s41408-024-01054-2
RAS mutations in myeloid malignancies: revisiting old questions with novel insights and therapeutic perspectives
Abstract
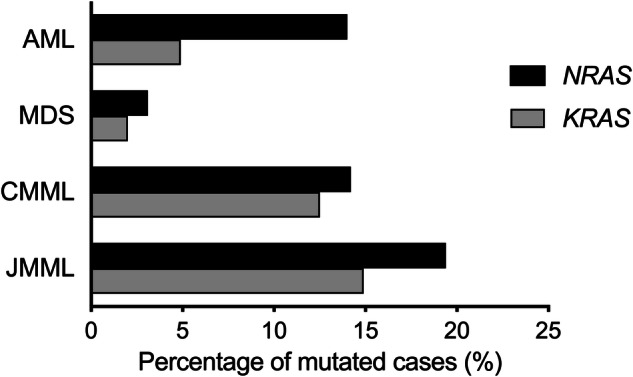
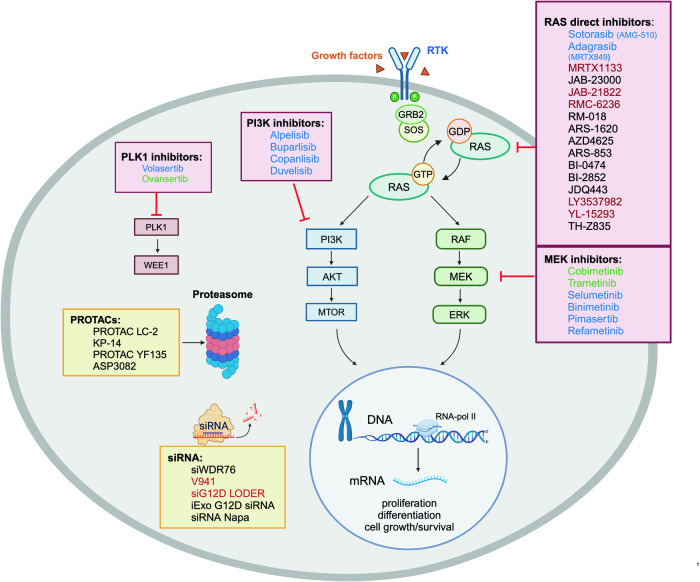
NRAS and KRAS activating point mutations are present in 10-30% of myeloid malignancies and are often associated with a proliferative phenotype. RAS mutations harbor allele-specific structural and biochemical properties depending on the hotspot mutation, contributing to variable biological consequences. Given their subclonal nature in most myeloid malignancies, their clonal architecture, and patterns of cooperativity with other driver genetic alterations may potentially have a direct, causal influence on the prognosis and treatment of myeloid malignancies. RAS mutations overall tend to be associated with poor clinical outcome in both chronic and acute myeloid malignancies. Several recent prognostic scoring systems have incorporated RAS mutational status. While RAS mutations do not always act as independent prognostic factors, they significantly influence disease progression and survival. However, their clinical significance depends on the type of mutation, disease context, and treatment administered. Recent evidence also indicates that RAS mutations drive resistance to targeted therapies, particularly FLT3, IDH1/2, or JAK2 inhibitors, as well as the venetoclax-azacitidine combination. The investigation of novel therapeutic strategies and combinations that target multiple axes within the RAS pathway, encompassing both upstream and downstream components, is an active field of research. The success of direct RAS inhibitors in patients with solid tumors has brought renewed optimism that this progress will be translated to patients with hematologic malignancies. In this review, we highlight key insights on RAS mutations across myeloid malignancies from the past decade, including their prevalence and distribution, cooperative genetic events, clonal architecture and dynamics, prognostic implications, and therapeutic targeting.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures

References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
