

Clinical and molecular outcomes from the 5-Year natural history study of SSADH Deficiency, a model metabolic neurodevelopmental disorder
- PMID: 38658850
- PMCID: PMC11044349
- DOI: 10.1186/s11689-024-09538-9
Clinical and molecular outcomes from the 5-Year natural history study of SSADH Deficiency, a model metabolic neurodevelopmental disorder
Abstract
Background: Succinic semialdehyde dehydrogenase deficiency (SSADHD) represents a model neurometabolic disease at the fulcrum of translational research within the Boston Children's Hospital Intellectual and Developmental Disabilities Research Centers (IDDRC), including the NIH-sponsored natural history study of clinical, neurophysiological, neuroimaging, and molecular markers, patient-derived induced pluripotent stem cells (iPSC) characterization, and development of a murine model for tightly regulated, cell-specific gene therapy.
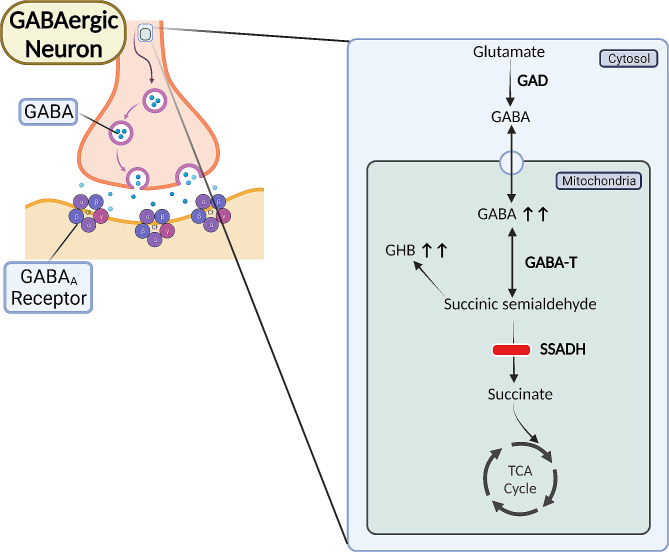
Methods: SSADHD subjects underwent clinical evaluations, neuropsychological assessments, biochemical quantification of γ-aminobutyrate (GABA) and related metabolites, electroencephalography (standard and high density), magnetoencephalography, transcranial magnetic stimulation, magnetic resonance imaging and spectroscopy, and genetic tests. This was parallel to laboratory molecular investigations of in vitro GABAergic neurons derived from induced human pluripotent stem cells (hiPSCs) of SSADHD subjects and biochemical analyses performed on a versatile murine model that uses an inducible and reversible rescue strategy allowing on-demand and cell-specific gene therapy.
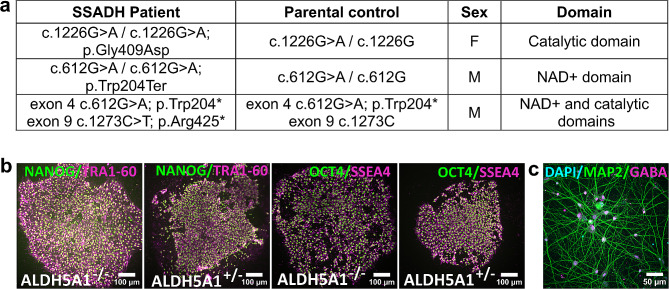
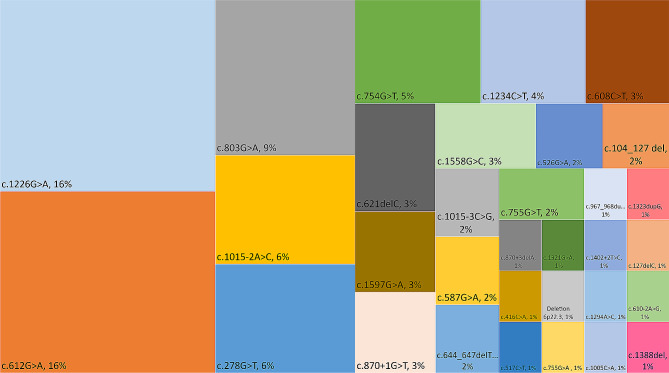
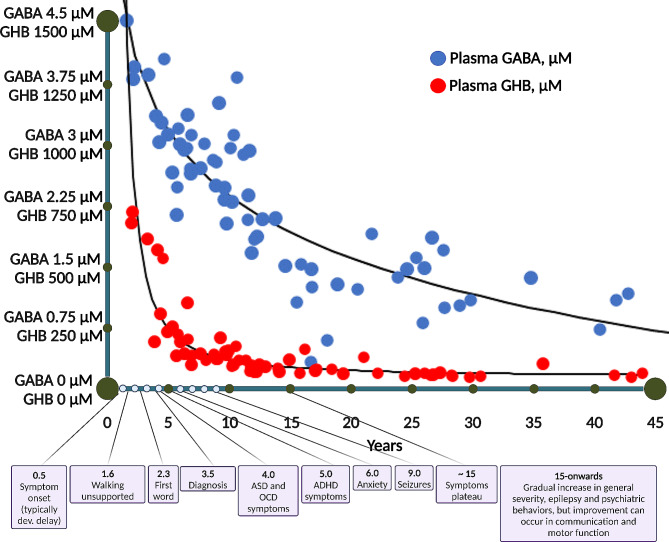
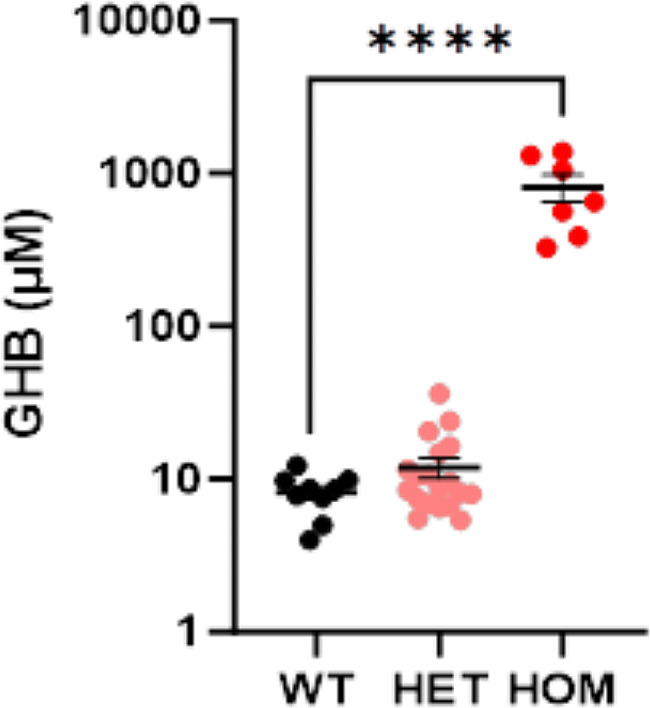
Results: The 62 SSADHD subjects [53% females, median (IQR) age of 9.6 (5.4-14.5) years] included in the study had a reported symptom onset at ∼ 6 months and were diagnosed at a median age of 4 years. Language developmental delays were more prominent than motor. Autism, epilepsy, movement disorders, sleep disturbances, and various psychiatric behaviors constituted the core of the disorder's clinical phenotype. Lower clinical severity scores, indicating worst severity, coincided with older age (R= -0.302, p = 0.03), as well as age-adjusted lower values of plasma γ-aminobutyrate (GABA) (R = 0.337, p = 0.02) and γ-hydroxybutyrate (GHB) (R = 0.360, p = 0.05). While epilepsy and psychiatric behaviors increase in severity with age, communication abilities and motor function tend to improve. iPSCs, which were differentiated into GABAergic neurons, represent the first in vitro neuronal model of SSADHD and express the neuronal marker microtubule-associated protein 2 (MAP2), as well as GABA. GABA-metabolism in induced GABAergic neurons could be reversed using CRISPR correction of the pathogenic variants or mRNA transfection and SSADHD iPSCs were associated with excessive glutamatergic activity and related synaptic excitation.
Conclusions: Findings from the SSADHD Natural History Study converge with iPSC and animal model work focused on a common disorder within our IDDRC, deepening our knowledge of the pathophysiology and longitudinal clinical course of a complex neurodevelopmental disorder. This further enables the identification of biomarkers and changes throughout development that will be essential for upcoming targeted trials of enzyme replacement and gene therapy.
Trial registration: ClinicalTrials.gov NCT03758521.
Keywords: Development; GABA; Neurotransmitters; Succinic semialdehyde dehydrogenase.
© 2024. The Author(s).
Conflict of interest statement
The authors AR and HHCL are co-founders and have equity in Galibra Neuroscience, Inc., which develops treatments for SSADHD including gene replacement therapy discussed in this study.
Figures
References
-
- Martin K, McConnell A, Elsea SH. Assessing prevalence and carrier frequency of Succinic Semialdehyde Dehydrogenase Deficiency. J Child Neurol. 2021;36:1218–22. - PubMed
-
- Jakobs C, Bojasch M, Mönch E, Rating D, Siemes H, Hanefeld F. Urinary excretion of gamma-hydroxybutyric acid in a patient with neurological abnormalities. The probability of a new inborn error of metabolism. Clin Chim Acta. 1981;111:169–78. - PubMed
-
- Gibson KM, Christensen E, Jakobs C, Fowler B, Clarke MA, Hammersen G, Raab K, Kobori J, Moosa A, Vollmer B. The clinical phenotype of succinic semialdehyde dehydrogenase deficiency (4-hydroxybutyric aciduria): case reports of 23 new patients. Pediatrics. 1997;99:567–74. - PubMed
-
- Rahbeeni Z, Ozand P, Rashed M, Gascon G, Al Nasser M, Al Odaib A, Amoudi M, Nester M, Al Garawi S, Brismar J. 4-Hydroxybutyric aciduria. Brain Dev. 1994;16:64–71. - PubMed
-
- Pattarelli PP, Nyhan WL, Gibson KM. Oxidation of [U-14 C] succinic semialdehyde in cultured human lymphoblasts: measurement of residual succinic semialdehyde dehydrogenase activity in 11 patients with 4-hydroxybutyric aciduria. Pediatr Res. 1988;24:455–60. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
