

This is a preprint.
Development of a First-in-Class RIPK1 Degrader to Enhance Antitumor Immunity
- PMID: 38659866
- PMCID: PMC11042424
- DOI: 10.21203/rs.3.rs-4156736/v1
Development of a First-in-Class RIPK1 Degrader to Enhance Antitumor Immunity
Update in
-
Development of a RIPK1 degrader to enhance antitumor immunity.Nat Commun. 2024 Dec 16;15(1):10683. doi: 10.1038/s41467-024-55006-2. Nat Commun. 2024. PMID: 39681571 Free PMC article.
Abstract
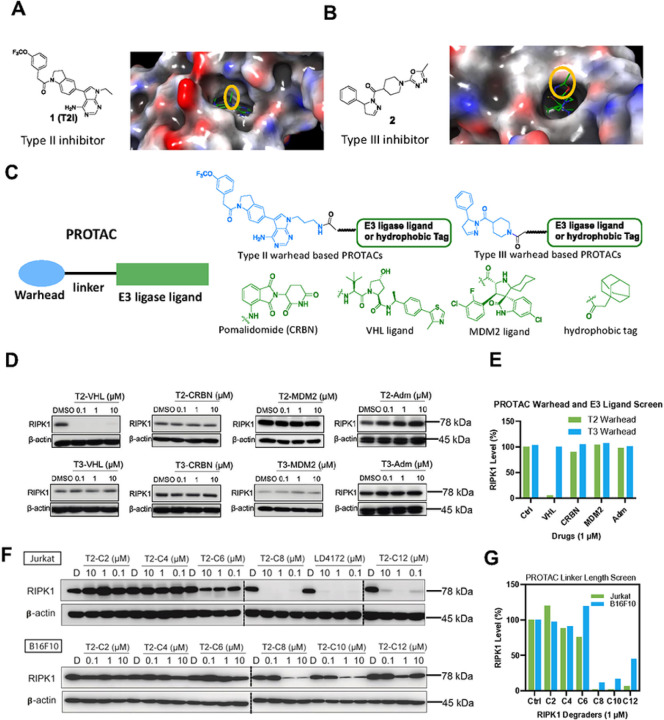
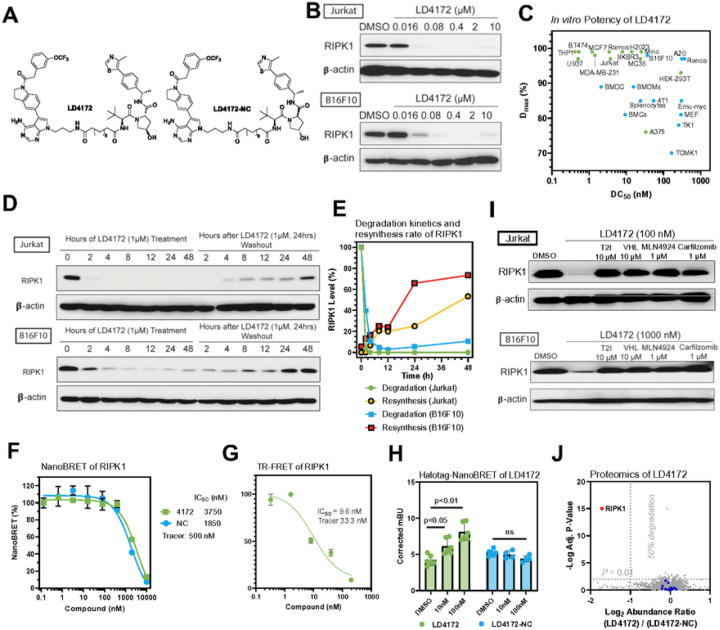
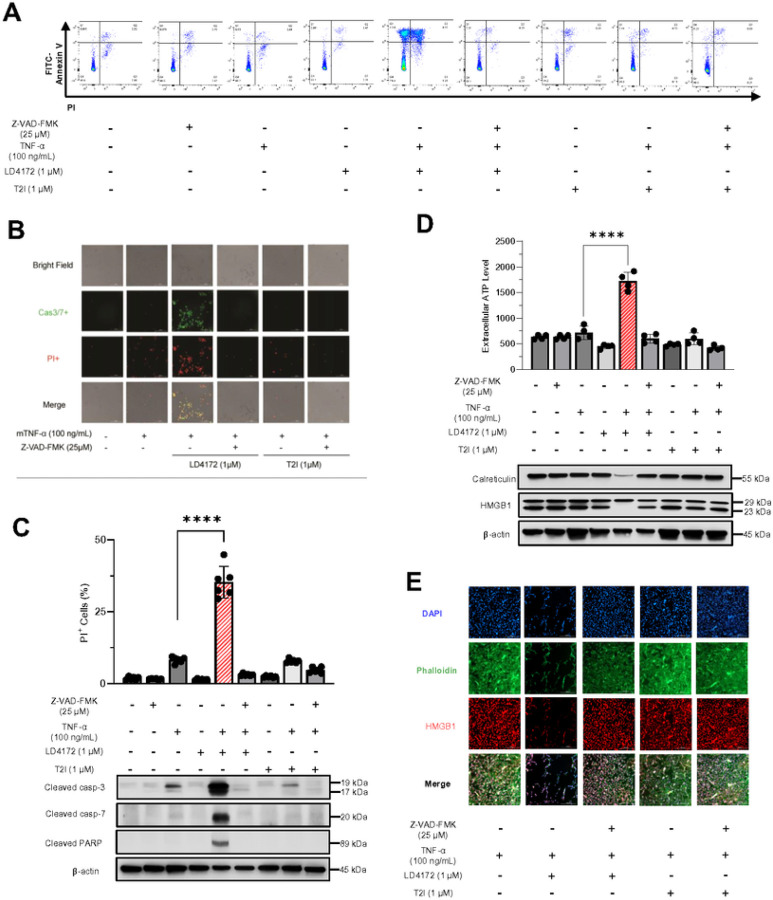
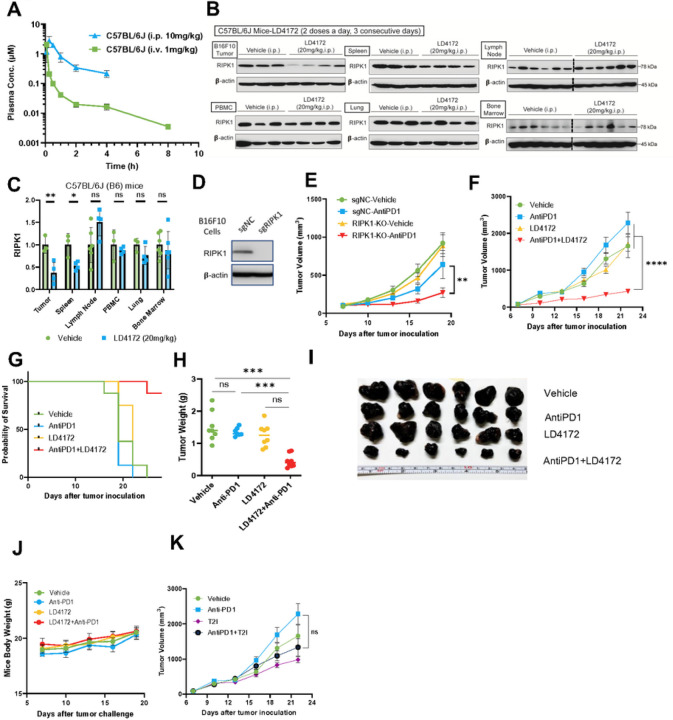
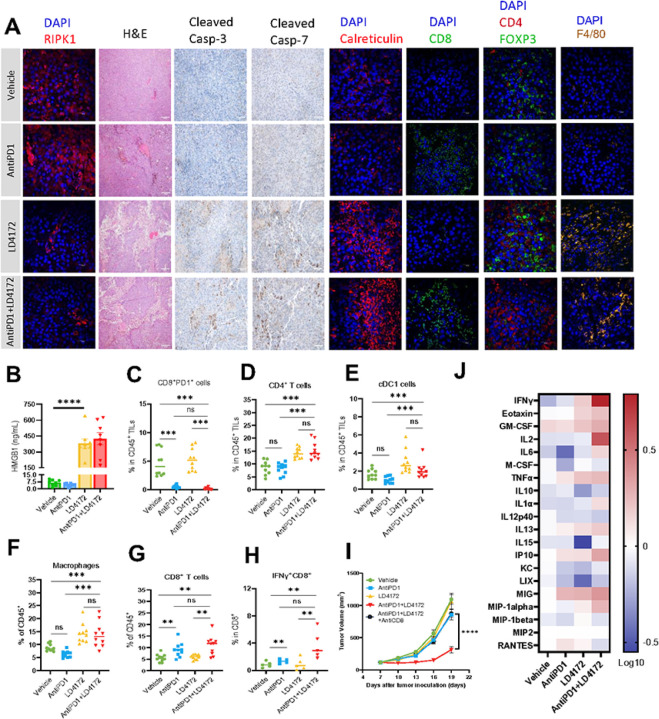
The scaffolding function of receptor interacting protein kinase 1 (RIPK1) confers intrinsic and extrinsic resistance to immune checkpoint blockades (ICBs) and has emerged as a promising target for improving cancer immunotherapies. To address the challenge posed by a poorly defined binding pocket within the intermediate domain, we harnessed proteolysis targeting chimera (PROTAC) technology to develop a first-in-class RIPK1 degrader, LD4172. LD4172 exhibited potent and selective RIPK1 degradation both in vitro and in vivo. Degradation of RIPK1 by LD4172 triggered immunogenic cell death (ICD) and enriched tumor-infiltrating lymphocytes and substantially sensitized the tumors to anti-PD1 therapy. This work reports the first RIPK1 degrader that serves as a chemical probe for investigating the scaffolding functions of RIPK1 and as a potential therapeutic agent to enhance tumor responses to immune checkpoint blockade therapy.
Conflict of interest statement
Conflicts of interest J.W. is the co-founder of CoActigon Inc. and Chemical Biology Probes, LLC. Y.X., D.L., and J.W. are inventors of a patent covering RIPK1 degraders reported in this work.
Figures
References
-
- Korman AJ, Garrett-Thomson SC, Lonberg N (2022) The foundations of immune checkpoint blockade and the ipilimumab approval decennial. Nat Rev Drug Discov 21:509–528 - PubMed
-
- Vesely MD, Zhang T, Chen L (2022) Resistance Mechanisms to Anti-PD Cancer Immunotherapy. Annu Rev Immunol 40:45–74 - PubMed
-
- Upadhaya S, Neftelinov ST, Hodge J, Campbell J (2022) Challenges and opportunities in the PD1/PDL1 inhibitor clinical trial landscape. Nat Rev Drug Discovery 21:482–483 - PubMed
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
