

Clinical and genetic studies for a cohort of patients with Leber congenital amaurosis
- PMID: 38662103
- PMCID: PMC11377616
- DOI: 10.1007/s00417-024-06450-9
Clinical and genetic studies for a cohort of patients with Leber congenital amaurosis
Abstract
Purpose: Leber congenital amaurosis (LCA) is a group of early-onset retinal degenerative disorders, resulting in blindness in children. This study aimed to describe the clinical and genetic characteristics of a cohort of patients with LCA and to investigate the retinal vascular characteristics in LCA patients.
Methods: Fifty-two children with LCA were included in the study. All patients underwent detailed ocular examinations. Electroretinography (ERG) was used to evaluate the retinal function. Optical coherence tomography (OCT) was used to assess the structure change of the retina for those patients who were able to cooperate very well. Panel-based next-generation sequencing was performed to identify pathogenic variants in genes associated with LCA. Diameters of the retinal vessels were measured using the EVision AI screening system with an artificial intelligence (AI) technique. An ultrasound Doppler was used to evaluate hemodynamic parameters, including peak systolic velocity (PSV), resistive index (RI), and pulsatility index (PI), in the ophthalmic, central retinal, posterior ciliary, carotid, and internal carotid as well as external carotid arteries in 12 patients aged from 3 to 14 years.
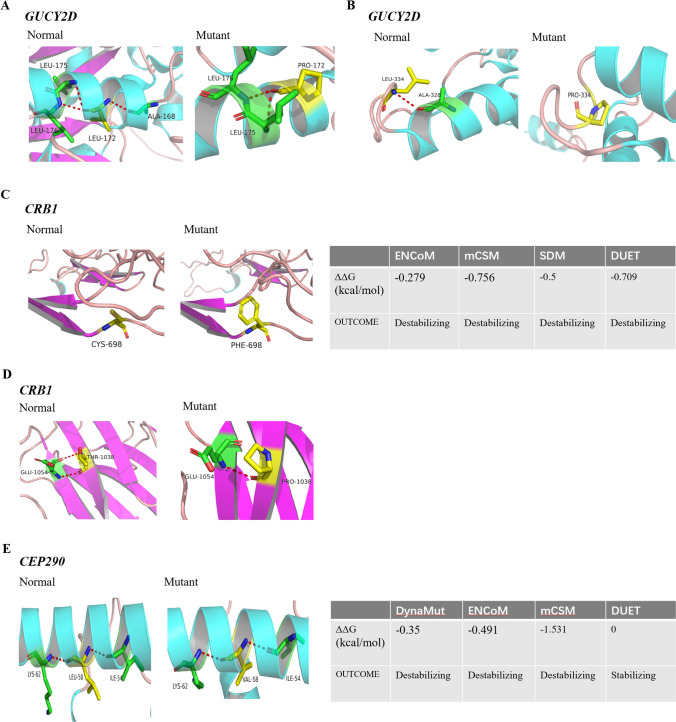
Results: We detected 75 pathogenic variants from ten genes of RPGRIP1, CEP290, GUCY2D, LCA5, AIPL1, CRB1, RPE65, CRX, RDH12, and TULP1, including 29 novel and 36 previously reported variants in 52 affected children with LCA, with the highest detective rate in RPGRIP1 (26.9%). Fundus appearance is diverse in patients with LCA, ranging from normal to severe peripheral or central retinopathy. Retinal vasculature was evaluated in 12 patients with different gene variants, showing narrowed arteries with an average diameter of 43.6 ± 3.8 μm compared to that of 51.7 ± 2.6 μm in the normal controls (P < 0.001, n = 12). Meanwhile, their hemodynamic parameters were changed as well in the ophthalmic artery (OA), with a decreased PSV (P = 0.0132, n = 12) and slightly increased PI (P = 0.0488, n = 12) compared to the normal controls. However, the hemodynamic parameters did not change significantly in the other vessels.
Conclusions: Blood supply to the eyeball is predicted to be reduced in patients with LCA, presumably due to photoreceptor cell degeneration. The novel identified variants will expand the spectrum of variants in LCA-related genes and be useful for studying the molecular mechanisms of LCA.
Keywords: Gene; LCA; Retinal vasculature.
© 2024. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures



Similar articles
-
Clinical and genetic characteristics of Leber congenital amaurosis with novel mutations in known genes based on a Chinese eastern coast Han population.Graefes Arch Clin Exp Ophthalmol. 2016 Nov;254(11):2227-2238. doi: 10.1007/s00417-016-3428-5. Epub 2016 Jul 16. Graefes Arch Clin Exp Ophthalmol. 2016. PMID: 27422788
-
Genetic and Clinical Profile of Retinopathies Due to Disease-Causing Variants in Leber Congenital Amaurosis (LCA)-Associated Genes in a Large German Cohort.Int J Mol Sci. 2023 May 17;24(10):8915. doi: 10.3390/ijms24108915. Int J Mol Sci. 2023. PMID: 37240262 Free PMC article.
-
Genetic and clinical findings in a Chinese cohort with Leber congenital amaurosis and early onset severe retinal dystrophy.Br J Ophthalmol. 2020 Jul;104(7):932-937. doi: 10.1136/bjophthalmol-2019-314281. Epub 2019 Oct 19. Br J Ophthalmol. 2020. PMID: 31630094
-
Leber Congenital Amaurosis – RETIRED CHAPTER, FOR HISTORICAL REFERENCE ONLY.2004 Jul 7 [updated 2013 May 2]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2025. 2004 Jul 7 [updated 2013 May 2]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2025. PMID: 20301475 Free Books & Documents. Review.
-
Leber congenital amaurosis: genes, proteins and disease mechanisms.Prog Retin Eye Res. 2008 Jul;27(4):391-419. doi: 10.1016/j.preteyeres.2008.05.003. Epub 2008 Jun 1. Prog Retin Eye Res. 2008. PMID: 18632300 Review.
Cited by
-
State of the Art on Inherited Retinal Dystrophies: Management and Molecular Genetics.J Clin Med. 2025 May 18;14(10):3526. doi: 10.3390/jcm14103526. J Clin Med. 2025. PMID: 40429522 Free PMC article. Review.
References
-
- Kumaran N, Moore AT, Weleber RG, Michaelides M (2017) Leber congenital amaurosis/early-onset severe retinal dystrophy: clinical features, molecular genetics and therapeutic interventions. Brit J Ophthalmol 101:1147–1154. 10.1136/bjophthalmol-2016-309975 10.1136/bjophthalmol-2016-309975 - DOI - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
