

Understanding and computational design of genetic circuits of metabolic networks
- PMID: 38662439
- PMCID: PMC11065555
- DOI: 10.1042/EBC20230045
Understanding and computational design of genetic circuits of metabolic networks
Abstract
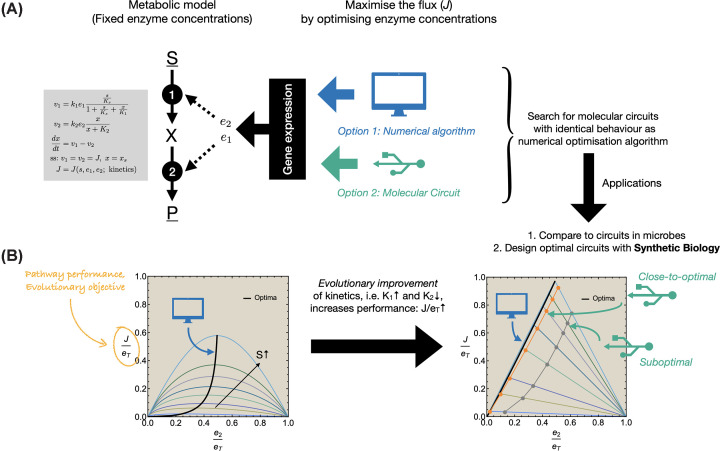
The expression of metabolic proteins is controlled by genetic circuits, matching metabolic demands and changing environmental conditions. Ideally, this regulation brings about a competitive level of metabolic fitness. Understanding how cells can achieve a robust (close-to-optimal) functioning of metabolism by appropriate control of gene expression aids synthetic biology by providing design criteria of synthetic circuits for biotechnological purposes. It also extends our understanding of the designs of genetic circuitry found in nature such as metabolite control of transcription factor activity, promoter architectures and transcription factor dependencies, and operon composition (in bacteria). Here, we review, explain and illustrate an approach that allows for the inference and design of genetic circuitry that steers metabolic networks to achieve a maximal flux per unit invested protein across dynamic conditions. We discuss how this approach and its understanding can be used to rationalize Escherichia coli's strategy to regulate the expression of its ribosomes and infer the design of circuitry controlling gene expression of amino-acid biosynthesis enzymes. The inferred regulation indeed resembles E. coli's circuits, suggesting that these have evolved to maximize amino-acid production fluxes per unit invested protein. We end by an outlook of the use of this approach in metabolic engineering applications.
Keywords: Escherichia coli; gene expression and regulation; mathematical modelling; microbiology.
© 2024 The Author(s).
Conflict of interest statement
The authors declare that there are no competing interests associated with the manuscript.
Figures



References
Publication types
MeSH terms
Grants and funding
- Centro de Investigación Científica y de Educación Superior de Ensenada, Baja California (CICESE)
- 713669/H2020 Marie Skłodowska-Curie Actions (MSCA)
- OCENW.XL21.XL21.007/Nederlandse Organisatie voor Wetenschappelijk Onderzoek (NWO)
- 613.009.131/Nederlandse Organisatie voor Wetenschappelijk Onderzoek (NWO)
LinkOut - more resources
Full Text Sources
Other Literature Sources