

Multireference Correlated Oscillator Strengths from Adiabatic Connection Approaches Based on Extended Random Phase Approximation
- PMID: 38669448
- PMCID: PMC11099974
- DOI: 10.1021/acs.jctc.4c00103
Multireference Correlated Oscillator Strengths from Adiabatic Connection Approaches Based on Extended Random Phase Approximation
Abstract
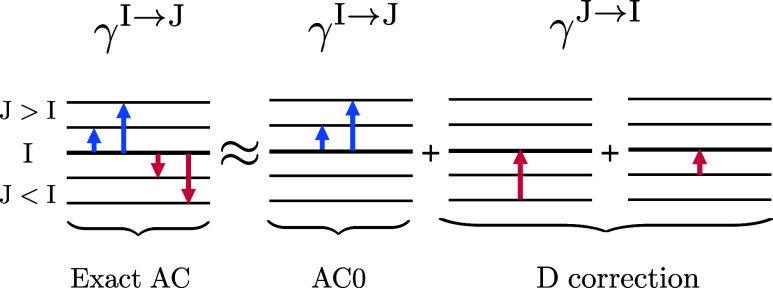
We show that accurate oscillator strengths can be obtained from adiabatic connection (AC) approaches based on the extended random phase approximation (ERPA) combined with multireference (complete active space, CAS) wave functions. The oscillator strengths calculated using the perturbation-corrected ERPA transition density matrices, proposed in this work, and the excitation energies calculated with recently introduced AC correlation energy methods, AC0 and AC0D, compete with accuracy in the perturbational CASPT2 approach and require less computational effort. AC0 and AC0D methods scale more favorably with the number of active orbitals than multiconfigurational perturbation approaches like CASPT2 and NEVPT2 thanks to their dependence on reduced density matrices up to the order of 2. Importantly, the newly developed approach for computing correlated transition dipole moments does not entail any additional costs, as all intermediate quantities become available when AC0 energies are being computed. We also test the performance of the recently proposed AC method corrected for the negative-transition contributions to the correlation energy, AC0D, for triplet excitation energies. Similarly, as for the singlet excitations, the correction improves the performance of the AC0 method, particularly for the low-lying excited states.
Conflict of interest statement
The authors declare no competing financial interest.
Figures



Similar articles
-
Correlation Energy from the Adiabatic Connection Formalism for Complete Active Space Wave Functions.J Chem Theory Comput. 2018 Jul 10;14(7):3493-3503. doi: 10.1021/acs.jctc.8b00213. Epub 2018 Jun 8. J Chem Theory Comput. 2018. PMID: 29787257
-
Excited states in the adiabatic connection fluctuation-dissipation theory: Recovering missing correlation energy from the negative part of the density response spectrum.J Chem Phys. 2021 Apr 28;154(16):164102. doi: 10.1063/5.0046852. J Chem Phys. 2021. PMID: 33940850
-
Electronic Excited States from the Adiabatic-Connection Formalism with Complete Active Space Wave Functions.J Phys Chem Lett. 2018 Sep 20;9(18):5534-5538. doi: 10.1021/acs.jpclett.8b02391. Epub 2018 Sep 11. J Phys Chem Lett. 2018. PMID: 30192553
-
Spinless formulation of linearized adiabatic connection approximation and its comparison with the second order N-electron valence state perturbation theory.Faraday Discuss. 2024 Nov 6;254(0):332-358. doi: 10.1039/d4fd00054d. Faraday Discuss. 2024. PMID: 39114978
-
Capturing the Dynamic Correlation for Arbitrary Spin-Symmetry CASSCF Reference with Adiabatic Connection Approaches: Insights into the Electronic Structure of the Tetramethyleneethane Diradical.J Phys Chem Lett. 2019 Aug 15;10(16):4668-4674. doi: 10.1021/acs.jpclett.9b01582. Epub 2019 Aug 5. J Phys Chem Lett. 2019. PMID: 31356083
References
-
- Toulouse J.; Gori-Giorgi P.; Savin A. A short-range correlation energy density functional with multi-determinantal reference. Theor. Chem. Acc. 2005, 114, 305–308. 10.1007/s00214-005-0688-2. - DOI
-
- Gritsenko O. V.; van Meer R.; Pernal K. Efficient evaluation of electron correlation along the bond-dissociation coordinate in the ground and excited ionic states with dynamic correlation suppression and enhancement functions of the on-top pair density. Phys. Rev. A 2018, 98, 062510.10.1103/PhysRevA.98.062510. - DOI
LinkOut - more resources
Full Text Sources