

TM7SF3 controls TEAD1 splicing to prevent MASH-induced liver fibrosis
- PMID: 38670107
- PMCID: PMC11113091
- DOI: 10.1016/j.cmet.2024.04.003
TM7SF3 controls TEAD1 splicing to prevent MASH-induced liver fibrosis
Abstract
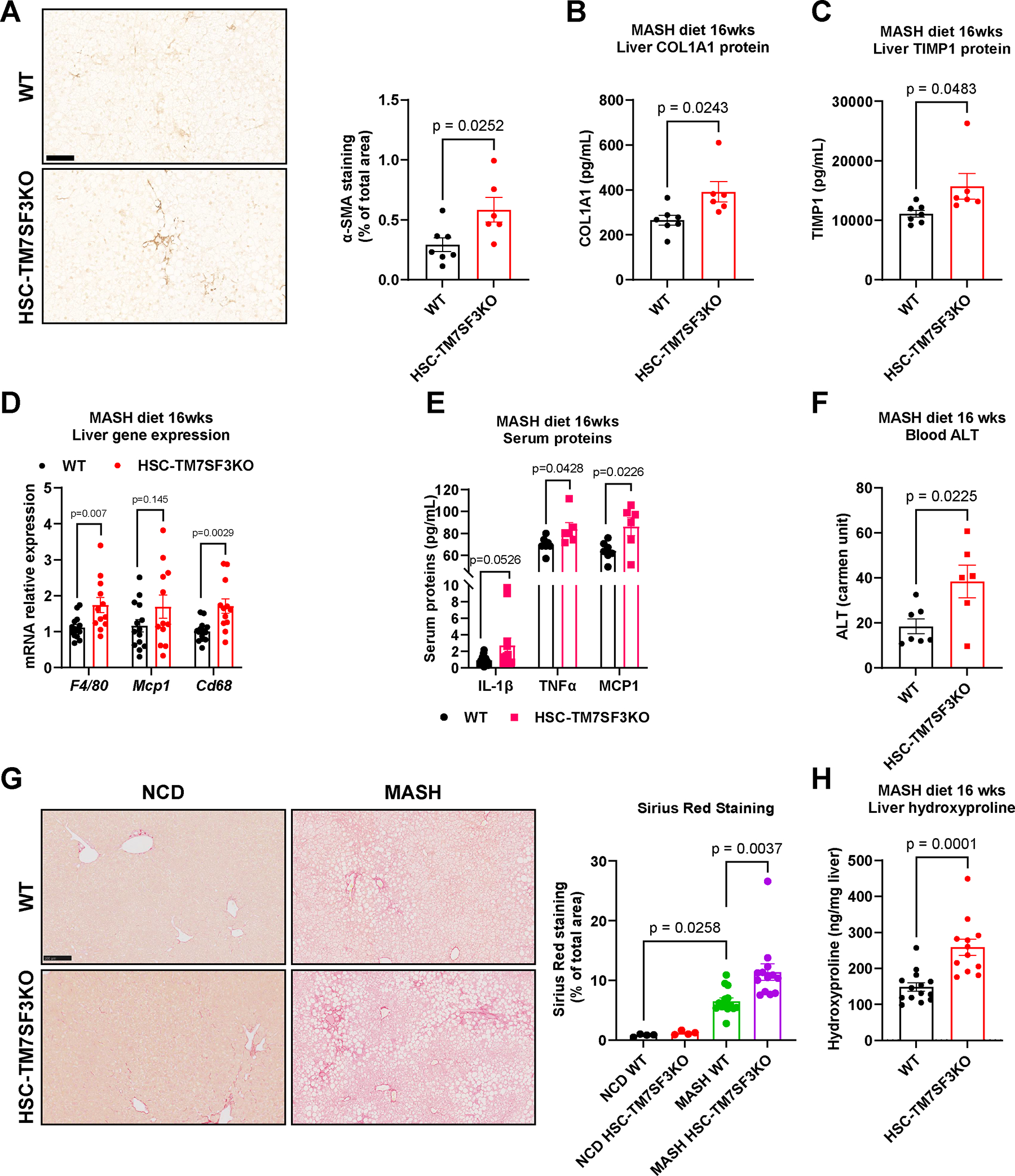
The mechanisms of hepatic stellate cell (HSC) activation and the development of liver fibrosis are not fully understood. Here, we show that deletion of a nuclear seven transmembrane protein, TM7SF3, accelerates HSC activation in liver organoids, primary human HSCs, and in vivo in metabolic-dysfunction-associated steatohepatitis (MASH) mice, leading to activation of the fibrogenic program and HSC proliferation. Thus, TM7SF3 knockdown promotes alternative splicing of the Hippo pathway transcription factor, TEAD1, by inhibiting the splicing factor heterogeneous nuclear ribonucleoprotein U (hnRNPU). This results in the exclusion of the inhibitory exon 5, generating a more active form of TEAD1 and triggering HSC activation. Furthermore, inhibiting TEAD1 alternative splicing with a specific antisense oligomer (ASO) deactivates HSCs in vitro and reduces MASH diet-induced liver fibrosis. In conclusion, by inhibiting TEAD1 alternative splicing, TM7SF3 plays a pivotal role in mitigating HSC activation and the progression of MASH-related fibrosis.
Keywords: ASO; Hippo pathway; MASH; NASH; TEAD1; TM7SF3; alternative splicing; fibrosis; hepatic stellate cells.
Copyright © 2024 Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests R.I. and J.M.O. are co-inventors on a provisional patent for the use of ASO 56 as an inhibitor of liver fibrosis.
Figures





References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
